
黏液水肿性苔藓(Lichen Myxedematosus)简称LM,又称丘疹性黏蛋白病(papular mucinosis)、硬化性黏液水肿(scleromyxoedema)、苔藓纤维黏蛋白病(lichen fibromucinodosis)或阿恩特-戈特尔综合征(Arndt-Gottron syndrome),是指在病理上真皮内有黏蛋白沉积和成纤维细胞增殖,临床上以局部或全身皮肤出现苔藓样丘疹、结节、斑块、硬皮病样改变等为特征的一种慢性进行性代谢性疾病。1953年由Montgomery和Underwood首次报道,为一罕见疾病。
病因及发病机制
病因不明。常伴有IgG型副蛋白血症,轻链主要是λ链,κ链很少,而IgM和IgA型副蛋白则少见。IgG型副蛋白是木瓜蛋白酶敏感的7S球蛋白,含较多赖氨酸,分子量约100kD(正常IgG为160kD),提示IgG球蛋白不完整,失去了有意义的抗原部分Fc片段。患者血浆在体外能刺激正常人成纤维细胞的DNA合成和增殖,若去除血浆中的IgG仍能引起成纤维细胞的增殖,说明细胞增殖是由血浆中的其他因子引起。也有报道IL-1、TNF-α和TNF-β能刺激真皮黏蛋白产生。早期有认为本病与浆细胞恶病质有关,但未得到证实。Earl认为本病是成纤维细胞和酸性黏多糖平衡失调所致。季素珍报道1例家族性黏液水肿性苔藓,提示本病可能与遗传有关。
临床症状
好发于中年人(30~50岁),无明显性别差异,无甲状腺病。皮疹主要为圆顶状、直径2~3mm、坚实的丘疹,肤色、淡红色或黄色,表面有蜡样光泽,丘疹数目不一,常局限,也可见于全身,密集成群,或融合成斑块,或呈线状、带状、串珠状、环状排列,在第一指指关节背侧可形成环形征(doughnut sign)。其他类型的皮疹有风团样斑块、结节、囊肿等,有时皮肤呈弥漫性浸润肥厚,类似硬皮病。好发于手背、指背、足背、前臂、大腿、上胸、背、腋部、面和颈部,分布对称。黏膜和头皮不受累。不痒或有微痒。有报道可出现同形反应。
根据临床、病理和是否有系统受累,Rongioletti等将黏液水肿性苔藓分为三型:硬化性黏液水肿或全身丘疹和硬皮病样型,局限性黏液水肿性苔藓或局限性丘疹黏蛋白病,不典型型或中间型。
硬化性黏液水肿
硬化性黏液水肿(scleromyxoedema)除丘疹和结节外,其主要特征是皮肤弥漫性浸润肥厚,呈硬皮病样改变,但能活动和捏起,手部受累可出现硬皮病样指端硬化。额部皮肤增厚最明显,眉间有嵴沟,鼻根部皮肤可肥厚凸起,使整个面部呈狮面样外观。硬皮病样损害严重时或晚期可使四肢和指(趾)弯曲受限,睁眼和张口困难等。
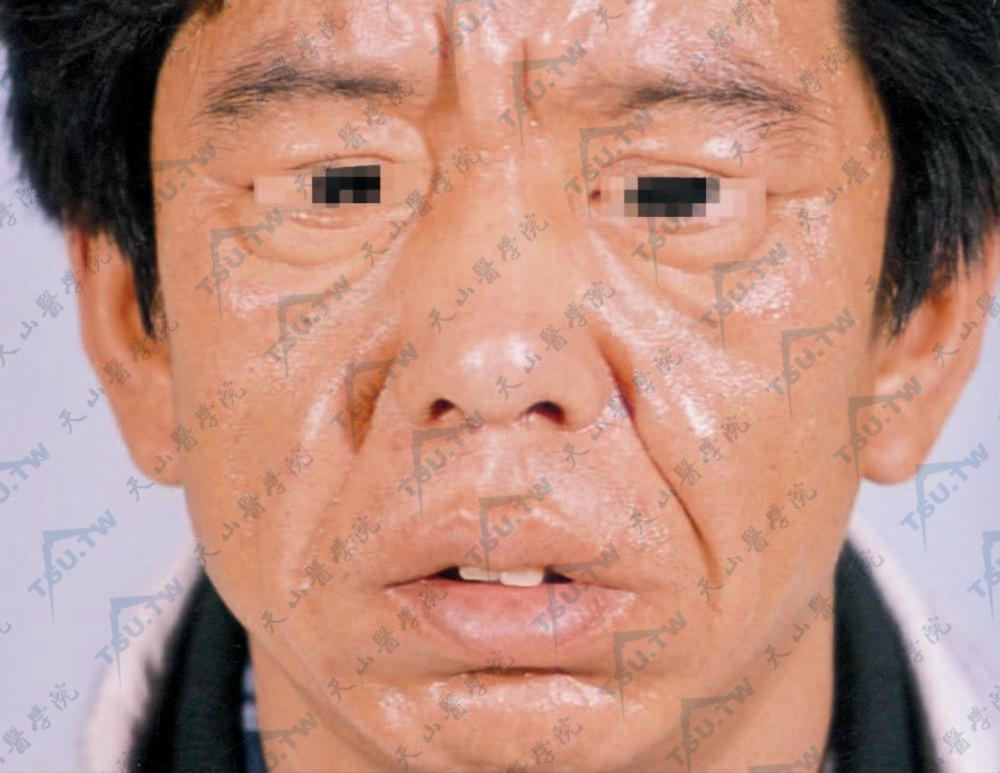
硬化性黏液水肿面部皮肤呈弥漫性浸润增厚,尤其在额部明显,眉间、鼻根部皮肤凸出肥厚。增厚的皮肤表面有苔藓样丘疹
此型常有多个系统受累,以副蛋白血症发生率最高(82.3%~90%),约10%单克隆γ病的患者可发展成多发性骨髓瘤。消化道受累最为常见,有食管蠕动消失、吞咽困难等;其次是呼吸系统,可出现呼吸困难、肺活量低下和肺动脉高压等;心脏病变约10%,表现为炎性肌病、动脉硬化(尤其是冠状动脉)、难控性高血压等;也可有肾脏受累、肝功能异常、消瘦、肥胖、声嘶等。累及中枢神经系统可发生进行性智力下降、头昏、意识模糊、构语障碍、癫痫发作,甚至出现晕厥和昏迷,约10%的患者有腕管综合征及其他外周神经病。若侵及肌肉可发生炎性肌病,有四肢肌无力。10%左右的患者有关节病变,表现为关节痛和游走性关节炎。8.8%的患者有雷诺现象,眼可出现角膜浑浊、眼睑闭合不全、睑外翻、脉络膜褶、视盘水肿等。本病可与盘状或系统性红斑狼疮、皮肌炎、硬斑病及系统性硬化等伴发。近年发现硬化性黏液水肿与艾滋病可能有关联。
局限性丘疹黏蛋白病
局限性丘疹黏蛋白病(localized popular mucinosis)其特点是无系统损害和副蛋白血症;皮疹以丘疹为主,也可有结节以及由丘疹融合成的斑块和结节,皮疹常局限在四肢和躯干。依据临床,本型再分为5个亚型:
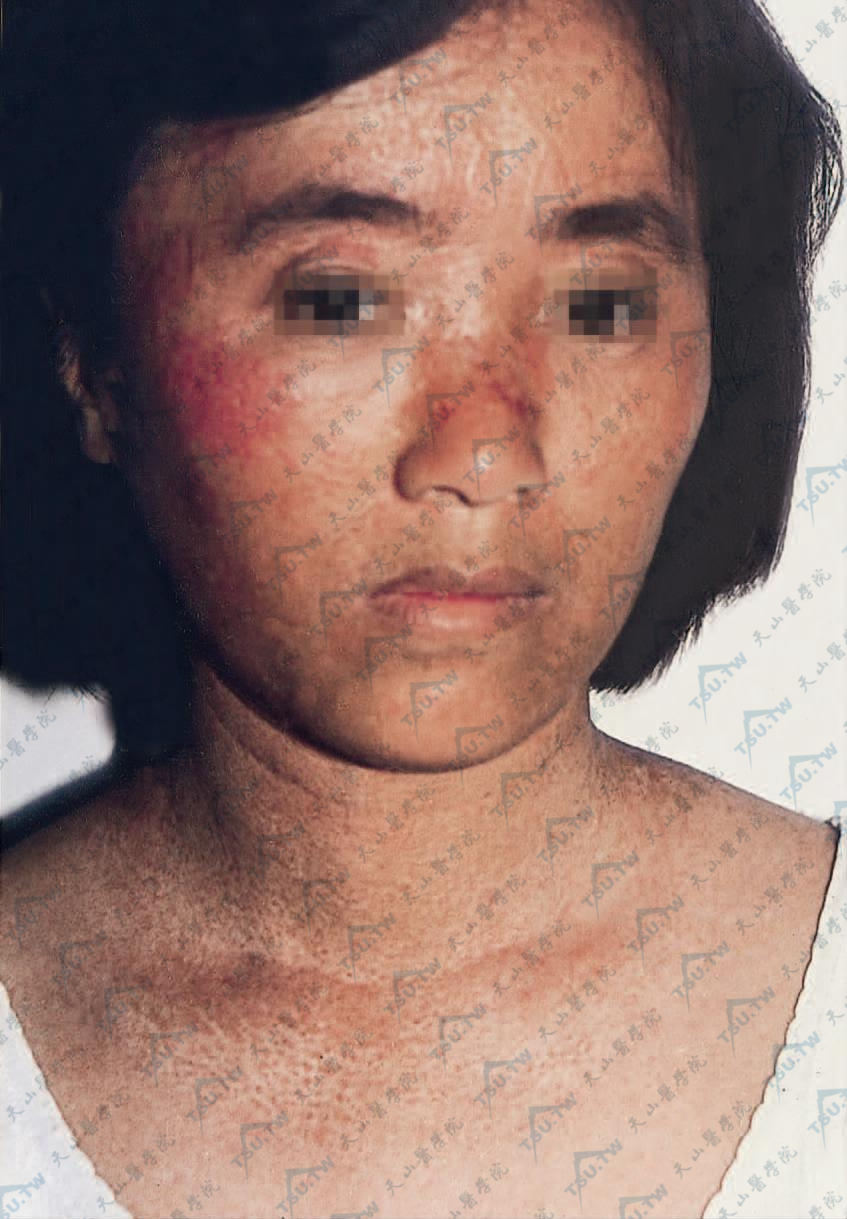
面、颈见淡红色或黄褐色苔藓样丘疹,密集成群
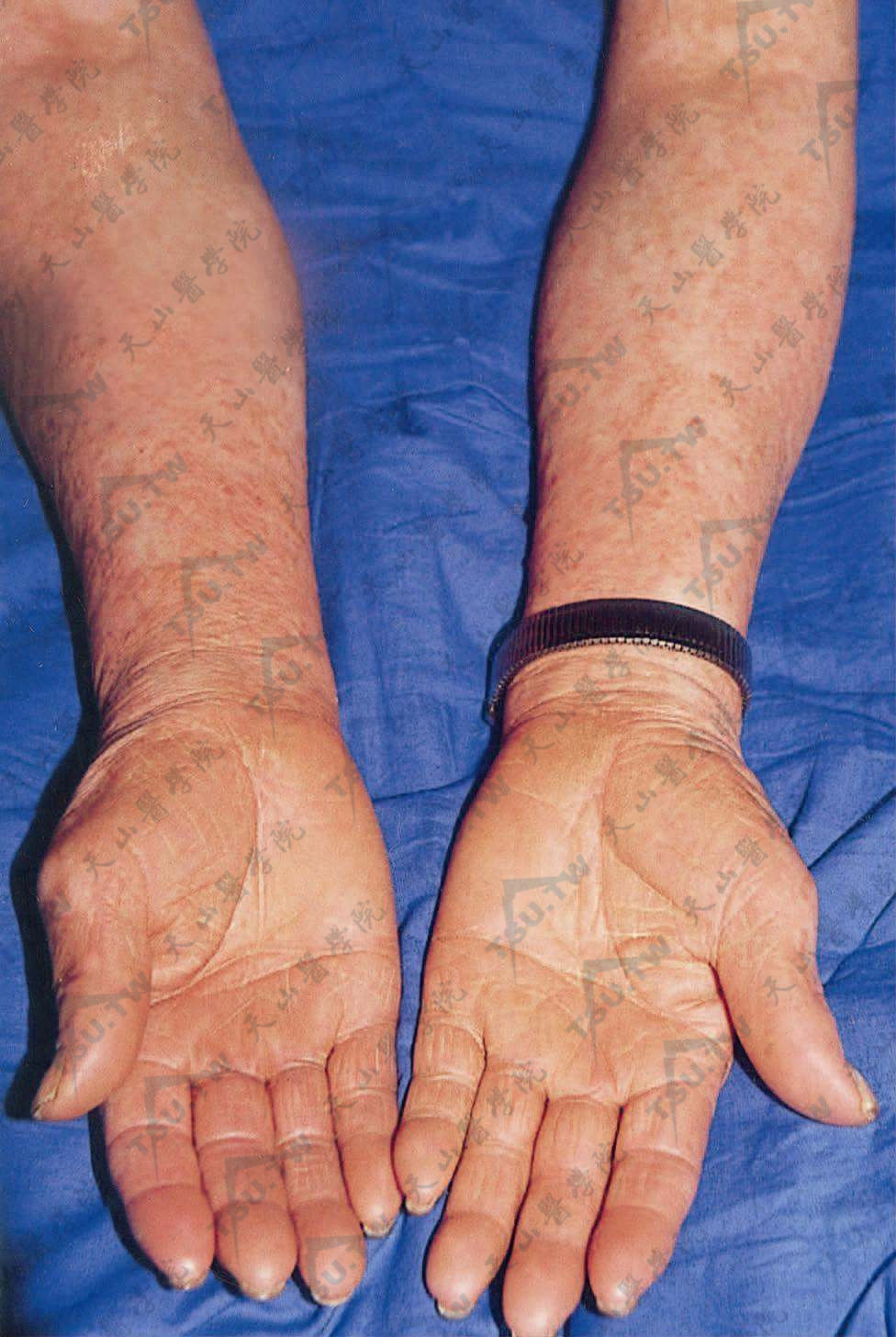
上肢见多数黄白色苔藓样丘疹,密集分布,有的成线状
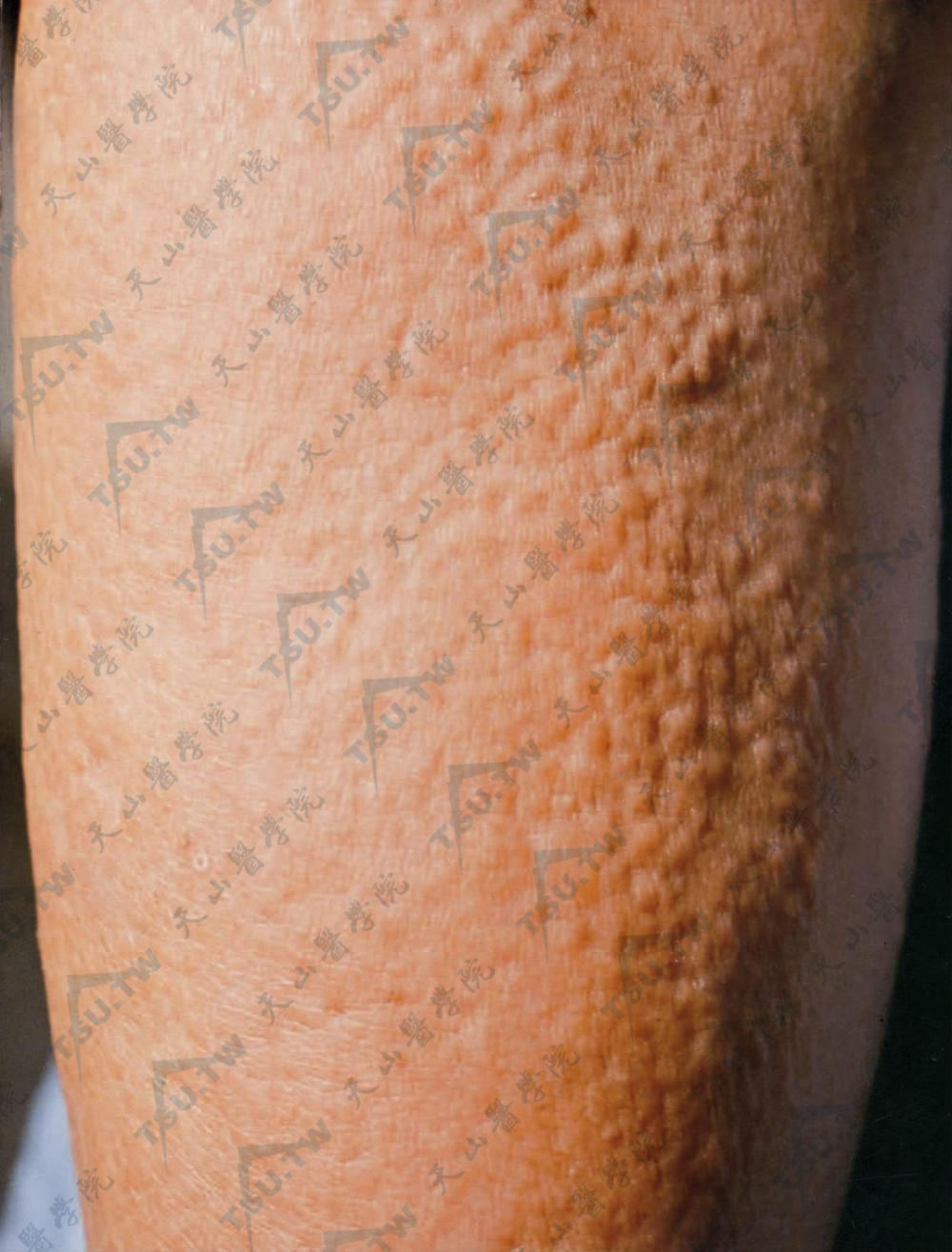
皮损为软的淡红或黄色苔藓样丘疹,可密集成群,有的排列成线状或斑片状,其下的皮肤呈浸润性增厚
- 散发丘疹性黏蛋白病(discrete popular LM):皮疹为2~5mm大,坚实光滑蜡样丘疹,肉色、红色或淡黄色,数个至数百个,孤立散在或融合成结节或斑块,好发于四肢和躯干,分布对称。疾病缓慢发展。
- 肢端持续性丘疹性黏蛋白病(acral persistent popular mucinosis):可能是一种独立的疾病。本型皮疹多发,丘疹呈象牙色或肉色,常见于手背和腕伸面,偶发于前臂远段。好发于女性,女男之比为4.7:1。皮疹持续,缓慢增多。该型和散发丘疹型可发生于HIV感染者。
- 自愈性丘疹性黏蛋白病(self-healing popular mucinosis):与自愈性青少年皮肤黏蛋白病可能是同一疾病。急性发疹,丘疹逐渐聚集成线状浸润性斑块,好发在面、颈、头皮、腹和股部,面和关节周围可有结节。患者可有发热、关节痛和无力。数周或数月后自愈。根据发病年龄再分青少年型和成人型。
- 婴儿丘疹黏蛋白病(popular mucinosis of infancy) 与婴儿皮肤黏蛋白病可能是同一疾病。皮疹为坚实的半透明丘疹,可有结节,发生在上臂(尤其肘部)和躯干。无系统受累,皮疹不会自行消退。
- 结节性黏液水肿性苔藓(nodular LM):皮疹为多发性结节,无丘疹或有少许丘疹,分布在四肢和躯干。
LM的不典型型
LM的不典型型(atypical forms of LM)症状不典型,介于两型之间。主要有4种:
- 硬化性黏液水肿,无单克隆γ病;
- 局限性LM伴单克隆γ病或系统受累;
- 局限性LM,具有各亚型的特征;
- 其他不典型病例。
本病总体预后较差,局限型一般预后良好,患者可长期存活,也有自愈者。合并系统病变者预后差,患者可死于非特异的并发症,如支气管肺炎、冠状动脉闭塞、恶性血液病等。
实验室检查:血沉增快,外周血嗜酸性粒细胞增多,血清白蛋白降低,黏蛋白增高,尿中酪氨酸增多,基础代谢率升高,甲状腺功能正常。血清IgG型副蛋白升高,轻链多是λ链,κ链很少,IgM和IgA型副蛋白少见。骨髓可有轻度浆细胞浸润,但放射学检查不能发现骨骼系统异常。
组织病理
最显著的变化是真皮上部胶原束间有大量黏蛋白沉积,pH为2.5时阿新蓝染色阳性,提示为酸性黏多糖。成纤维细胞和胶原增多,胶原束排列不规则。但局限型的成纤维细胞和胶原增生程度较低,无纤维化。若为硬化性黏液水肿,黏蛋白弥漫性沉积在真皮网状层的上、中部,成纤维细胞显著增生,胶原增多,有纤维化,弹性纤维断裂和减少,毛囊可有萎缩。血管和附属器周围有淋巴细胞和一些组织细胞、多形核细胞和浆细胞浸润。胶原束排列紊乱,许多区域胶原束被黏蛋白分裂成单一胶原纤维。表皮变化轻微,可正常或因黏蛋白挤压而变薄。
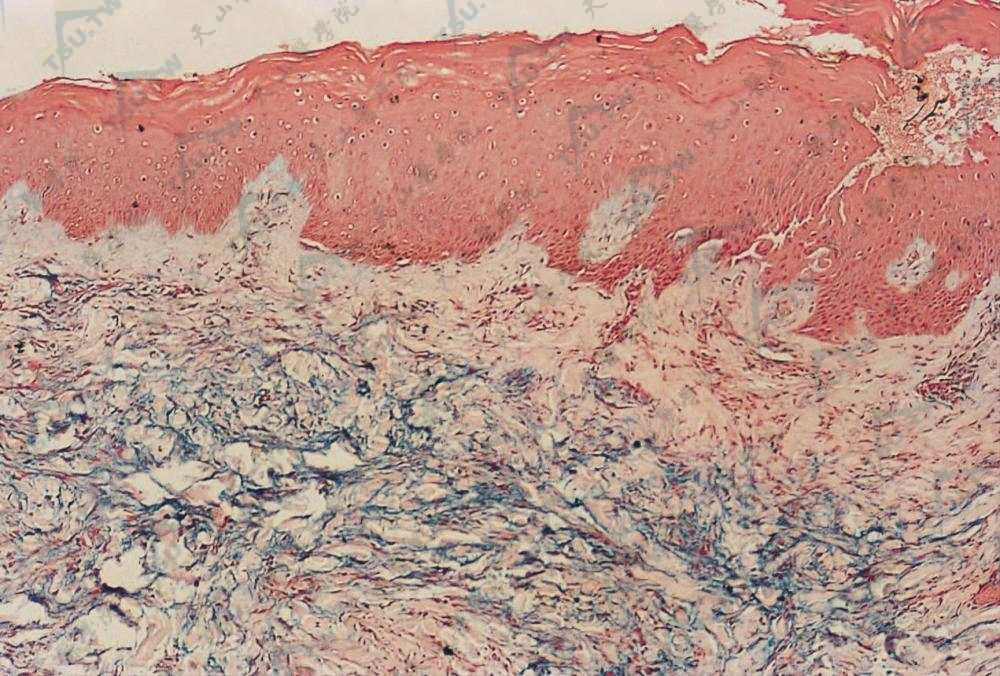
真皮中部黏液沉着,淡蓝色(阿新蓝染色×200)
电子显微镜观察示成纤维细胞有长的胞质突起,粗面内质网扩张,有许多细的胶原纤维,表明是幼稚的胶原纤维。
肌肉可发生非典型坏死性肌病,表现为肌纤维坏死、严重的Ⅱ型纤维萎缩和空泡变。在肾、心、肾上腺、胰和肾乳头的血管外膜可有黏蛋白沉积。
诊断及鉴别
依据临床表现,结合组织病理、系统检查和血清单克隆球蛋白的检测等可诊断和分型。但需注意:
- 发生在陈旧性瘢痕上或周围的浸润性皮疹与瘢痕性结节病相似;
- 手背和耳部的丘疹应与环状肉芽肿鉴别;
- 与系统性硬皮病的鉴别在于本病皮肤有丘疹,增厚变硬的皮肤能捏起和推动,而硬皮病无丘疹,变硬的皮肤既不能捏起,也不能移动;
- 根据本病有丘疹易与硬肿病区别。
预防及治疗
目前尚无特效疗法。
局部治疗:外涂或皮损内注射糖皮质激素有效,也可局部注射玻璃酸,或试用电子束、浅层X线、PUVA等。
系统治疗:主要用于硬化性黏液水肿,尤其是重者。大剂量糖皮质激素可暂时性抑制内脏病变的进展(中剂量无效),化疗药常选用美法仑(melphalan)、环磷酰胺、环孢素、盐酸苯丁酸氮芥等。美法仑小剂量长期使用,3个月后可获效,但需注意其潜在的毒副作用。Howsden等每天用环磷酰胺200mg(逐渐减量至50mg),共6个月,皮肤恢复到正常。糖皮质激素和化疗药联合应用有望提高疗效。异维A酸和阿维A酯对皮损有效,但对系统损害的疗效还不清楚。血浆置换结合糖皮质激素冲击治疗或(和)免疫抑制剂治疗可能有效。迄今已报道有效的疗法有静脉滴注免疫球蛋白(IVIg)、沙利度胺、2-氯脱氧腺苷、干扰素等;Righi等用IVIg[每月,2g/(kg·5d)]治疗3例硬化性黏液水肿获得成功。
饮食疗法:Saez-Rodriguez等报道2例局限型肥胖患者,仅给予低热能饮食(5020.8J/d),随着体重的减轻,皮疹逐级消退。对毁容性损害,整容外科和皮肤磨削术可有帮助。
