
本病简称P0H,是皮肤骨化的一种罕见类型,1994年被首次报道。其特征是婴儿期皮肤和软组织进行性异位骨化。
病因及发病机制
病因不明。与Albright遗传性骨营养不良相似,系由来自父系不活动的GNAS1基因突变所致,该基因负责产生鸟嘌呤核苷酸结合蛋白(G蛋白)a刺激活性多肽。骨化可能是由内外多因素所致。内因有局部组织效应(如细胞密度、组织的血管分布、pH、P02及钙、磷和枸橼酸盐离子的浓度)、某些酶(如碱性磷酸酶)、某些诱导物或生长因子、遗传素质(genetic endowment)、分化和发育。异位骨化产生同皮肤骨瘤。患者血清碱性磷酸酶、乳酸脱氢酶和磷酸肌酸激酶升高,提示有骨形成增加和肌肉损伤。
临床症状
常发生在生后6个月内,但多在1个月时发病,女性多于男性。皮疹多为P0H的第一表现。初起为谷粒样小丘疹,有沙样的硬度,以后可出现较大的丘疹和结节,可融合成大的斑块。皮疹广泛,随机分布,也可为单侧性或仅累及某一个部位。皮疹发生前无损伤或炎症,创伤似乎不会加重皮损,但手术切除皮损后可复发。疾病呈进行性发展,无畸形,但可出现较严重的并发症如溃疡、感染、严重疼痛、关节强直、肢体长度不一等。
实验室检查:血清碱性磷酸酶、乳酸脱氢酶(LDH)、肌酸磷酸激酶(CK)可升高,血清钙、磷、PTH、1,25-二羟维生素队等正常。
组织病理
真皮和皮下组织内有异位骨(网状骨甚至成熟的膜内骨),也可有非骨化的钙沉着。活检应包括皮下组织,因真皮浅层可仅有钙化而无骨化。肌肉内也可有异位骨。
诊断及鉴别
根据临床表现,结合组织病理和影像学检查可诊断。与其他骨化疾病的鉴别见下表。
进行性骨发育异常与其他骨化疾病的鉴别
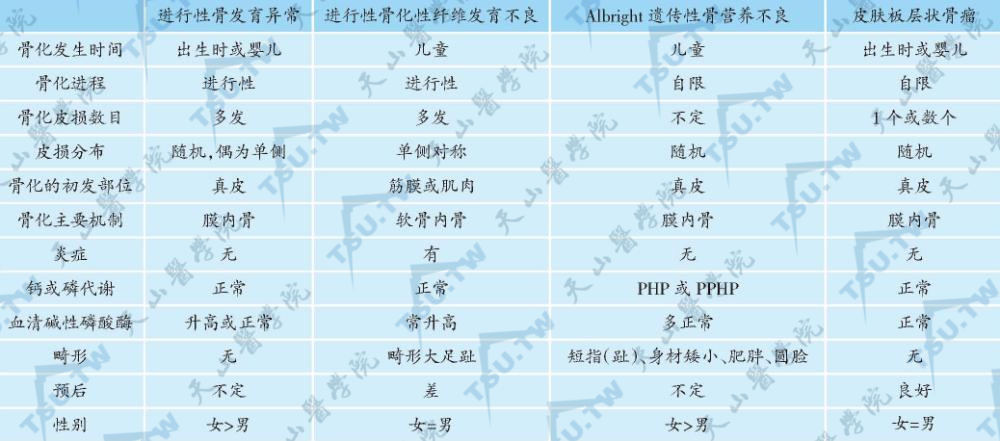
注:PHP——假性甲状旁腺功能减退症;PPHP——假假性甲状旁腺功能减退症。
预防及治疗
尚无有效治疗来阻止骨化的进展,异位骨可手术切除。
系统的医学参考与学习网站:天山医学院, 引用注明出处:https://www.tsu.tw/edu/2494.html
