
着色性干皮病(Xeroderma Pigmentosa、简称XP)是一种常染色体隐性遗传性皮肤病,发病率约1:25万。两性发病相当。父母常为近亲结婚。患者对日光高度敏感,有畏光现象。光暴露部位皮肤萎缩、大量的雀斑样色素加深斑,继而出现新生物,可有多系统累及,许多患者可伴有眼球、神经系统等病变。
病因及发病机制
最早由Morutz Kaposi等于1870年命名,是第一个与DNA损伤修复缺陷有关的人类疾病,可累及各种族人群。患者细胞存在UV照射后DNA损伤修复功能缺陷,患者的皮肤部位缺乏核酸内切酶,不能修复被紫外线损伤的皮肤的DNA,因此在日光照射后皮肤容易被紫外线损伤,先是出现皮肤炎症,继而可发生皮肤癌。患者发生皮肤癌的可能性几乎是100%。
着色性干皮病并非一个独立疾病。通过成纤维细胞融合技术并监测DNA修复,目前分为8种亚型,7个互补组(A-G)和1个变异型(XPV)。变异型最轻,皮肤肿瘤发生较晚(40岁左右发生),很少有神经系统异常,C组、E组较轻,其他组较重。
- XPA组:最为严重,约占XP的25%,是位于9q34.1的XPA基因突变所致。XPA基因是一种锌指蛋白,能够与受损DNA特异性结合。
- XPB基因(又称ERCC3),属于螺旋酶,位于2号染色体q21,起DNA螺旋酶和ATP酶的作用。
- XPC基因位于染色体3p25.1,编码DNA结合蛋白,对基因组非转录区修复是必需的,在损伤识别的第一步起关键作用。
- XPD基因(又称ERCC2)代表另一种螺旋酶,位于染色体19q13.2。XPD蛋白具有5′-3′解链酶活性,参与损伤DNA的解链。
- XPE基因突变导致典型野生型,位于染色体11q12-q13和11q11-p12,编码二聚体蛋白,参与损伤DNA的识别。
- XPF基因(又称ERCC4),位于染色体16p13.3,这种类型罕见,主要见于日本人。XPF蛋白具5′核酸内切酶活性,参与损伤DNA的剪切。
- XPG组罕见,XPG基因(又称ERCC5)位于染色体13q32-33。XPG蛋白具3′核酸内切酶活性,参与损伤DNA的剪切。
- XPV基因(又称POLH),位于染色体6p21.1-6p12,编码DNA聚合酶,可以绕过损伤DNA合成完整的子链DNA。
近日,在XP皮肤肿瘤患者中,证实有活化癌基因N-ras,Ha-ras和C-myc。这些与UV导致的DNA损伤有关。
临床症状
(一)皮肤病变
出生时皮肤正常,一般在出生后6个月至3岁发病。但大多数患者在20岁前即进入肿瘤期。有些患者发展缓慢,但也有一些在几年内即发生许多肿瘤。初期的皮损发生在曝光部位,光敏感最为常见,日晒部位发生水疱、大量雀斑、伴有色素减退和萎缩、皮肤干燥、毛细血管扩张、瘢痕形成和日光角化病。雀斑淡至暗棕色,针头至1cm以上大小,可互相融合而形成不规则的色素沉着斑片,最初入冬可见其色较淡,其后即持久不退,其间逐渐夹杂有毛细血管扩张及小血管瘤。常见疣状角化,可自行消退或恶化。严重的慢性光化性损伤使皮肤呈异色病样外观。可在前3~4年即出现第一个恶变的肿瘤,多为基底细胞癌,数量可很多,有时可为着色性。鳞癌也常见,黑素瘤也不少见,且为多发性,可因广泛转移而早年死亡。XP患者发生皮肤肿瘤的风险是正常人的1000倍。肿瘤发展的平均时间为8年。最易发生在面部、颈部和头部。有时尽管组织学上有恶变但病程慢,甚至可活至成年期。其他纤维肉瘤和血管肉瘤少见。
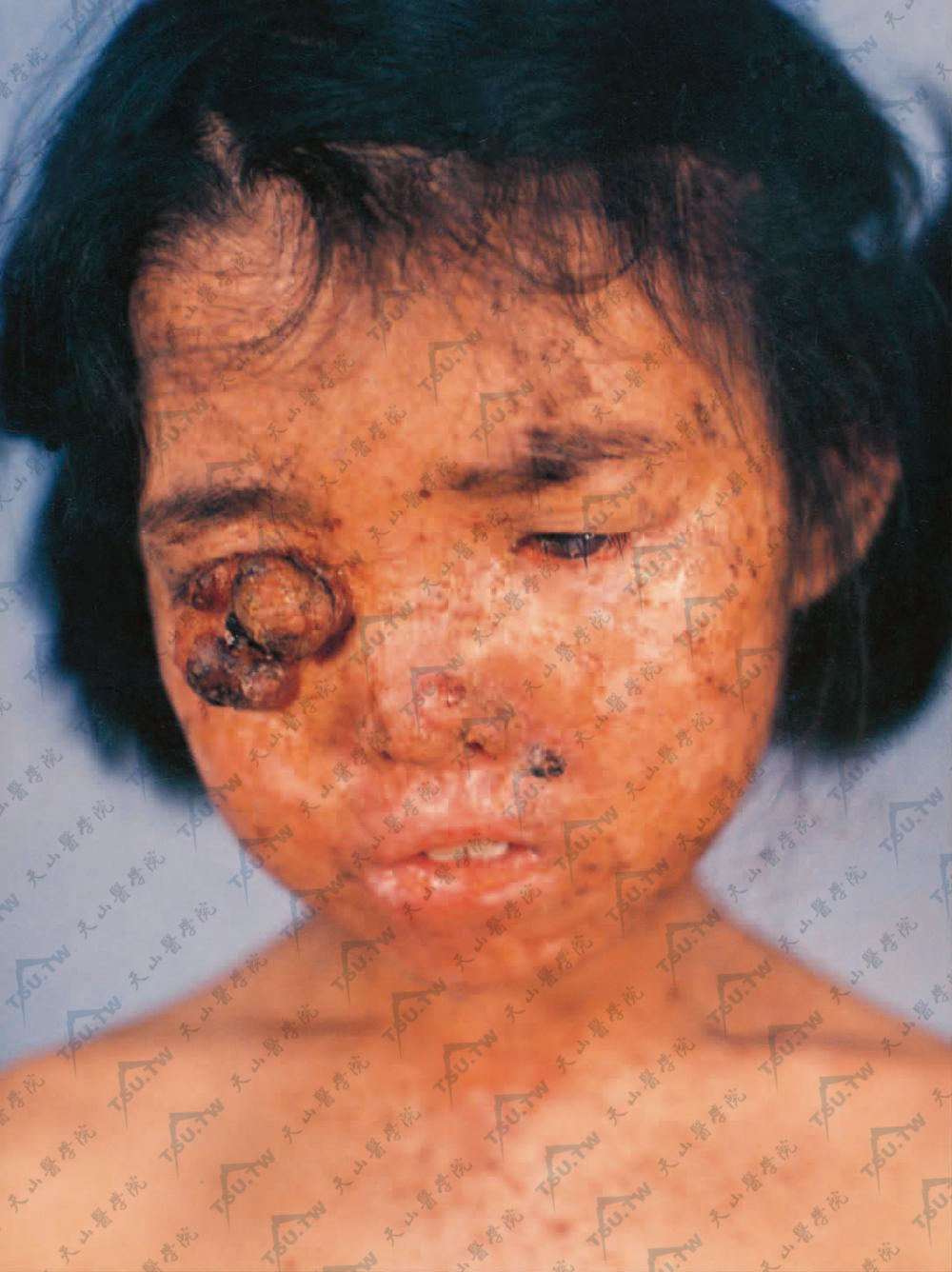
上图:面部皮肤干燥,出现雀斑,暗棕色针头至豆大,数目多,互相融合成不规则色素斑。其间夹杂有毛细血管扩张、白色萎缩斑点和疣状角化。右眼部发生恶性肿瘤
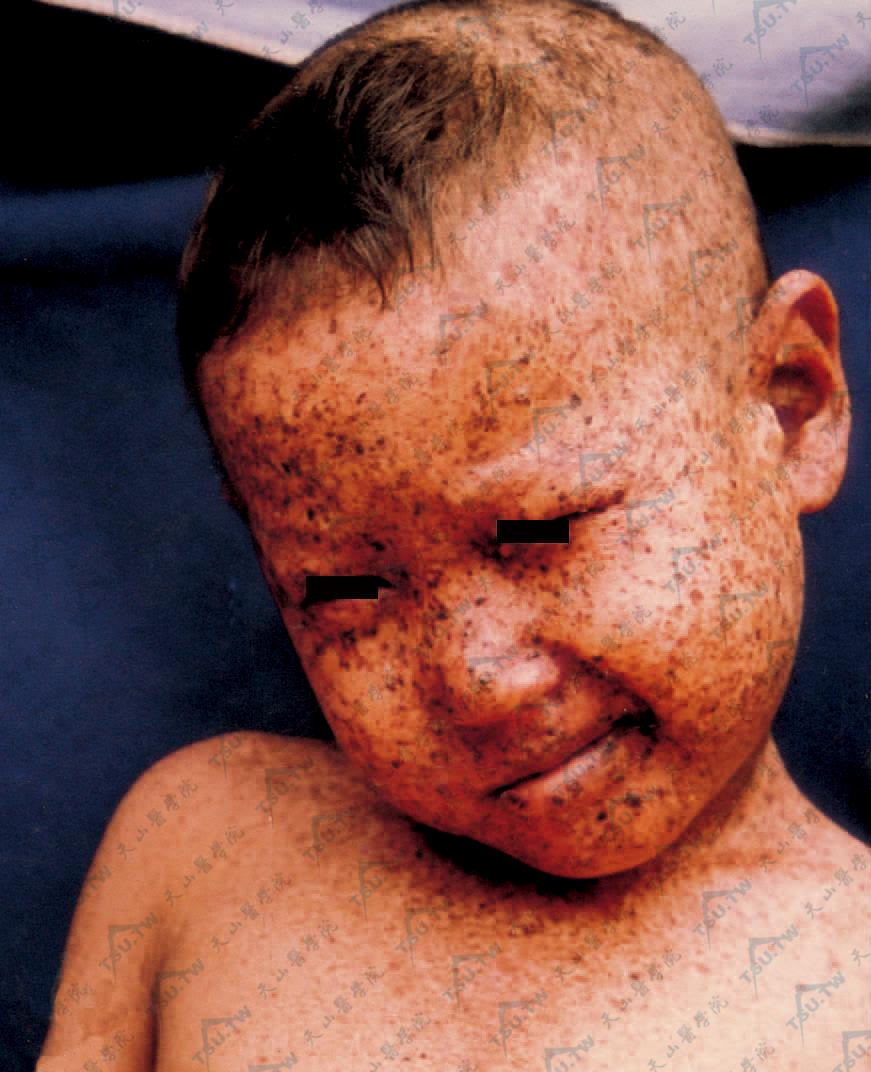
头、面、颈见多数雀斑、疣状角化、白色萎缩斑点及毛细血管扩张,有眼损害,畏光
毛发及指甲常正常,但齿可有缺陷。本病常在10岁前死亡,2/3患者于20岁前死亡。鳞癌及黑素瘤广泛转移是死亡原因之一。许多患者常因易发生感染而死亡。轻症或适当治疗的患者有时可活过中年。
(二)眼部损害
见于80%的患者。包括畏光性结膜炎、睫毛缺失、角膜溃疡、瘢痕形成和穿孔、眼睑外翻和内翻,以及发生在眼睑部位的鳞状细胞癌基底细胞癌和黑素瘤。XPA组眼部损害更多见。
(三)神经系统改变
可累及18%的患者,常见小头、智力障碍、舞蹈手足徐动症、小脑性共济失调和感觉神经性耳聋等。最严重的神经系统症状是De Sanctis-Cacchione综合征(XPA组),C组、E组和XP其他亚型不发生神经疾病。
(四)其他
口腔结构的严重萎缩和癌症亦可能系暴露于日光中所致,可有张口困难,舌尖部可出现毛细血管扩张和其他病变,如鳞癌。内脏恶性肿瘤的危险性比正常人群高10~20倍。
组织病理
早期病理变化为非特异性,可有角化过度,马尔匹基层变薄,表皮突部分萎缩,部分伸长,基底细胞层黑素不规则集聚,黑素细胞正常或增多。中期的病理变化似日光性角化病,角化过度与色素增深更加明显。表皮部分萎缩,部分棘层肥厚,甚至呈非典型性向下生长。表皮细胞核排列紊乱。真皮上部胶原纤维嗜碱性变和日光性弹性纤维病样改变。真皮内有较多的黑素颗粒及噬黑素细胞。到晚期则表现为各种肿瘤的组织改变。
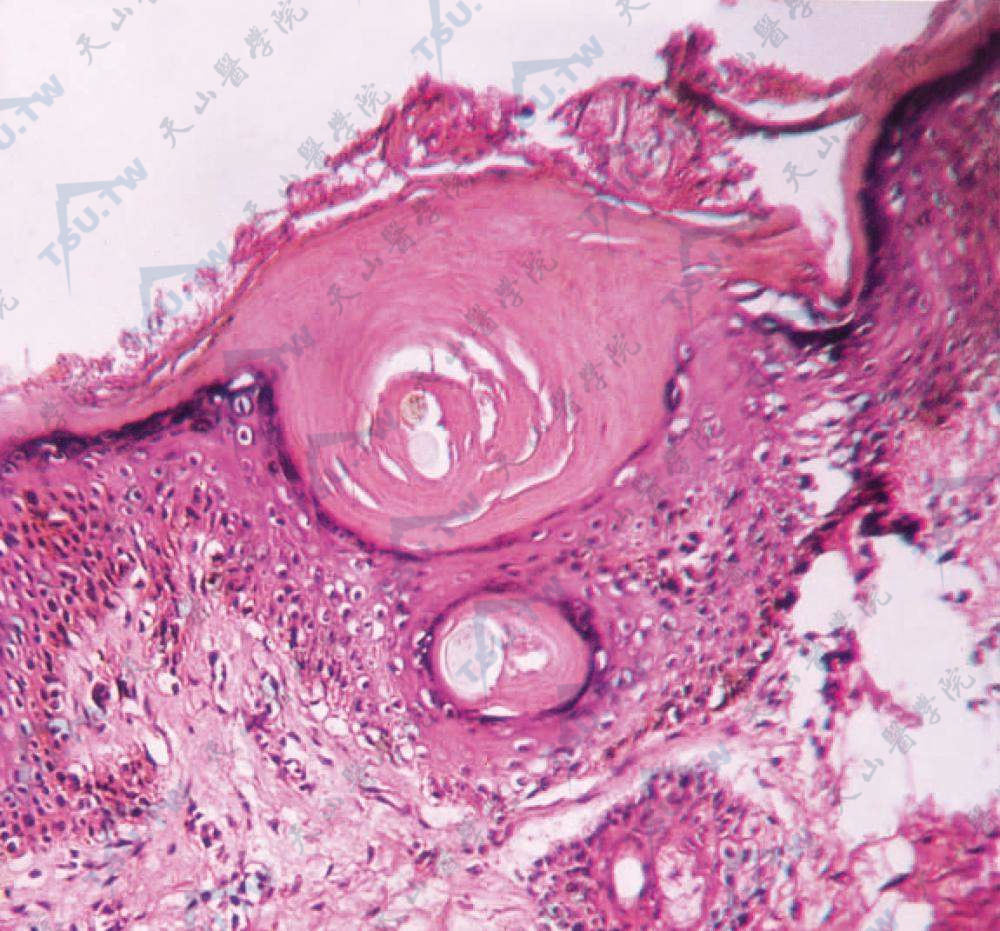
角化过度,毛囊角质栓,基底层色素增加(HE染色×400)
诊断及鉴别
典型病例,根据临床表现即可确诊。但发病较晚者应与着色性干皮病样综合征相鉴别。后者要到30~40岁才发病,表皮内DNA修复复制过程正常,但紫外线照后DNA合成较差。
早期或轻症皮损应与雀斑相鉴别,还应与罗斯蒙德-汤姆森综合征(Rothmund-Thomson syndrome)、Petuz-Jeher综合征及科凯恩综合征(Cockayne syndrome)相鉴别。
预防及治疗
一旦确诊,尽量避免日晒并使用遮光剂保护皮肤,如25%二氧化钛霜和5%PABA液。尽早切除肿瘤,面部损害切除后有时应进行整形植皮。患者家属应仔细检查,以便及早发现轻症患者进行保护和预防。
近年研究发现芳香维A酸,每日0.2~0.5mg/kg,口服可有效地减少皮肤肿瘤形成。另外,目前新的治疗方法是用T4内切核酸酶V能够特异识别环丁烷嘧啶二聚体,可用于着色性干皮病的酶替代治疗。
