
本病又名帕皮永-勒菲弗综合征(Papollin-Lèfèvre syndrome),为一种少见的常染色体隐性遗传性疾病。由编码溶酶体组织蛋白酶的基因突变引起,最近报道本病与Capthasin-C基因突变有关。发病率估计为(1~4)/1000000。本病表现为掌跖角化过度和牙周变性。一般在出生后数月内发生,但也可在出乳牙后发病。常见的皮肤改变有掌跖的界限清楚的红斑性过度角化性损害,可延伸到手足背侧。这些改变常在幼儿时开始出现,可发生于肘、膝和跟腱区域,可出现指甲横沟。在大多数病例,牙改变伴随着皮肤病变而出现,但有些病例皮肤病变在牙病变之前出现。病情一般冬重夏轻。
临床症状
本综合征有掌跖皮肤角化过度和牙周病,牙齿过早脱落,两者多同时起病,但也有先发生皮损者。部分患者的父母为近亲结婚。但皮损的严重程度和牙周感染之间并无相关性。
常于乳齿长出后(1~5岁)发病,在掌跖发生境界清楚的红斑性角化过度损害,伴鳞屑和皲裂,并逐步扩展至手足侧缘、手足背、足跟、踝部、肘与膝部,可出现严重的银屑病样损害,分布对称,冬季症状加重,常伴有手足多汗和臭汗症,足跖部尤重。毛发正常或稀疏,少数可有甲改变,表现为甲肥厚、弯曲或有横沟发生,还可伴发蜘蛛脚样指(趾)和沟纹舌。
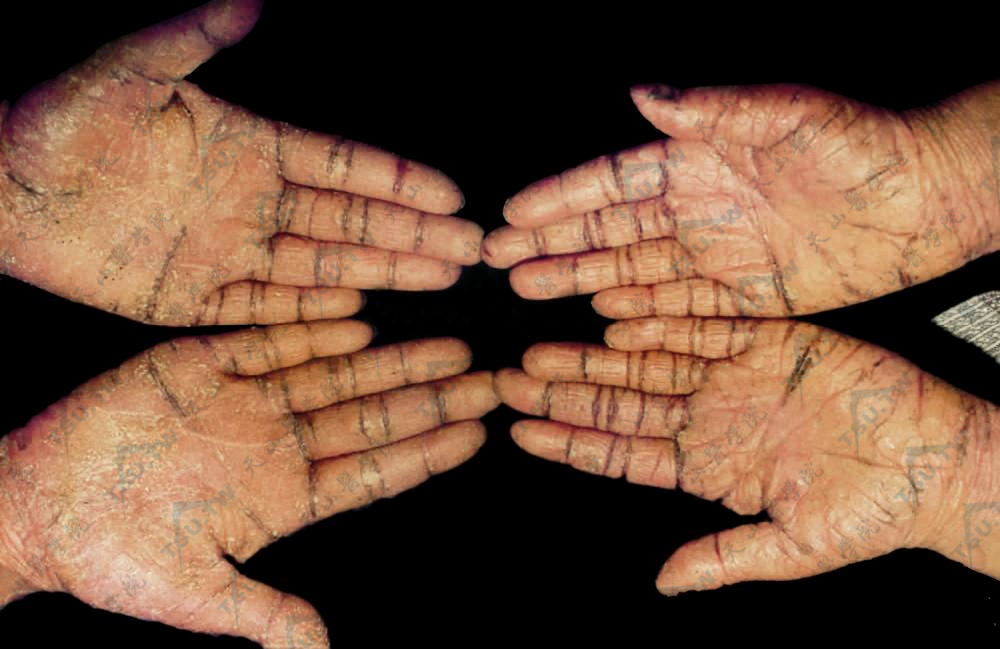
兄弟二人掌部呈境界清楚红斑性角化过度,伴鳞屑及皲裂
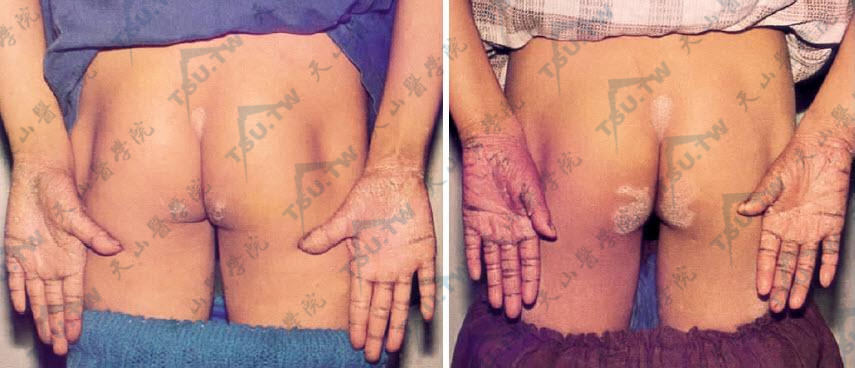
兄弟二人同样发病,两手掌及臀部、骶部均有红斑角化损害
牙周病的早期发作原因是由于多形核白细胞功能受损和改变。本病与肢端骨质疏松症和化脓性肝脓肿有联系。在脉络膜丛和小脑幕有无症状的异位性钙沉着。一般在皮疹初发的同时,出现乳齿的牙周变性,其表现较特殊,一般乳牙发育正常,但一长出牙周的支持组织即开始破坏,牙龈充血肿胀、溃烂而容易出血、牙周溢脓,有牙周袋形成和口臭,牙槽骨吸收、萎缩。乳牙多在4~5岁脱落,恒牙生长发育可正常,但可以同样方式过早脱落。牙周炎性退行性变伴暂时性牙齿的脱落是反复发生的,局部淋巴结炎普遍存在,而口腔其他组织均正常。治疗可延缓牙和皮肤的异常变化。本病如不予治疗,在10岁左右患儿的牙齿可全部脱落。
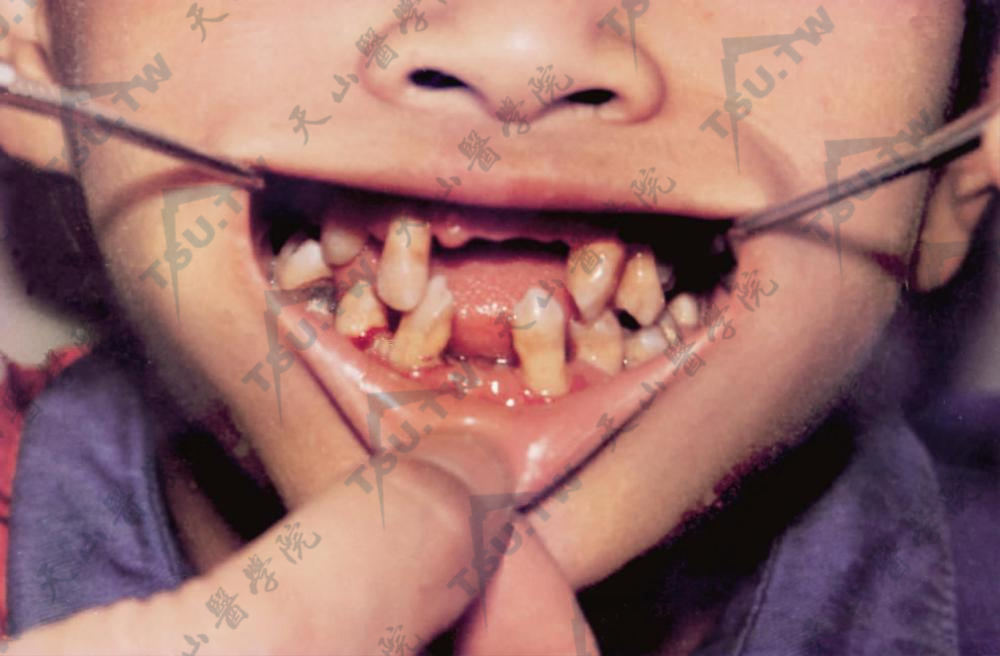
牙齿发育异常,牙龈充血肿胀、萎缩,牙齿脱落
此外,Haim曾报道一个家族中4例患者均有细长指,指端尖细并呈钩形;Bach等注意到患者在脉络膜丛和脑幕附着处有无炎症性钙化。
组织病理:与弥漫性掌跖角皮症同。
鉴别诊断
应与下列疾病相鉴别:
Meleda病:一般在出生时或婴儿期患病,掌跖部位弥漫性红斑、鳞屑和角化过度,有指甲改变和体格发育不良,但毛发、牙齿或眼无缺陷;
毛发红糠疹:虽有掌跖角化过度,但典型皮损为毛囊角化性丘疹融合成鳞屑状斑块,无牙齿改变。
预防及治疗
对皮损及牙周病作对症治疗;对牙龈炎引起的牙槽骨破坏,应及早拔除残牙及使用牙托。
Bergfeld等报道用阿维A酯治疗对皮损有效,但对牙周损害疗效不一;Nazzaro等用阿维A(Acitretin)治疗本病同样反应良好。上述治疗对患者的恒齿有保护作用,但最好在第一个恒齿萌出之前(约5.5岁时)就开始给药。
由于本病为常染色体隐性遗传,且致病基因已经明确,故应告诫患者避免近亲结婚。
)因本病病情发展快,故在发现幼年早发性掌跖角化过度的患儿时,应注意口腔情况,发现异常时应及时到口腔科诊治。
