
本病又称先天性脂肪萎缩性糖尿病(congenital lipoatrophic diabetes)、脂肪萎缩性糖尿病(lipoatrophic diabetes)或贝拉尔迪恩利综合征(Berardinelli syndrome)。为一种少见的先天性脂肪萎缩症,有家族史,表现为皮下和内脏脂肪萎缩、缺失,常伴有内脏疾患如肝大及其他先天性异常(骨生长快、高脂血症),后期有糖尿病。国内1987年报道1例。
病因及发病机制
病因不明,Brun Zell报道一家族有兄妹5人患病,认为本病为一常染色体隐性遗传性疾患,父母近亲结婚,具有综合征全部症状者为纯合子,而杂合子仅表现高脂血症。该病有Cllq13基因异常,患者有间脑或下丘脑-垂体系统功能紊乱,血中下丘脑释放的因子水平增高,有的病例下丘脑有病理性损害。
有1例患者用扭转下丘脑功能异常药治疗后,血中下丘脑释放因子消失,恢复皮下脂肪沉积,但其他患者没有获得同样疗效。也有认为患者胰岛素受体基因有缺陷,或储存脂肪有缺陷。
临床症状
出生时就有皮下脂肪和皮肤以外组织脂肪缺损。在婴儿及儿童期,骨骼生长过快,身高超过同龄人,肌肉组织增生,肌肉显露明显,具有男性型体型。外生殖器增大,伴阴蒂或阴茎增大,易被误诊为性早熟。肝脾大使腹部明显膨隆,常有脐疝。肝大禁食后可变小,可引起肝硬化、肝功能衰竭。青春期前后发生胰岛素抵抗性糖尿病,出现糖尿病性肾脏、视网膜、神经等病变。成年期身高超过预测高度。患者基础代谢率高,食欲亢进,全身性多汗。虽全身脂肪消失,但皮肤仍保持其固有弹性,坐立、走路均正常。有高甘油三酯血症,可发生皮肤发疹性黄瘤。
有泛发性多毛症,甚至出生时就有,头发多而弯曲,前发际几乎长到眉毛部位。有轻至中度精神发育迟缓、间隙性精神分裂症、偏瘫等。所有患者因面部脂肪缺损均有特征性憔悴面容。大部分头颅长,关节特别是手和足关节变大。有广泛性色素沉着,尤其腋下和腹股沟褶皱部位,可伴有线状表皮增厚,有黑棘皮病样外观。部分病例有肥厚性心肌病、周围性肺动脉狭窄、肾脏病变如非低补体性肾病综合征、肾肥大,患者不易怀孕,成人常死于糖尿病并发症或肝、心脏病变。
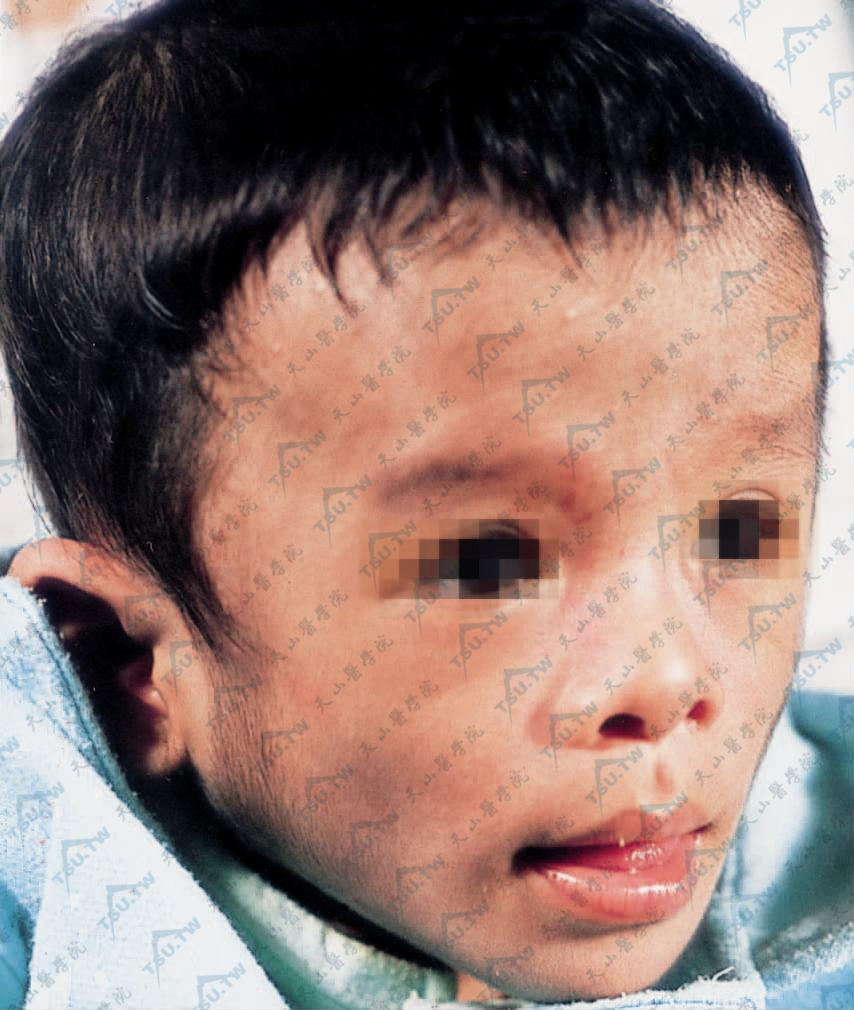
患儿呈“早老貌”,面颊部凹缩
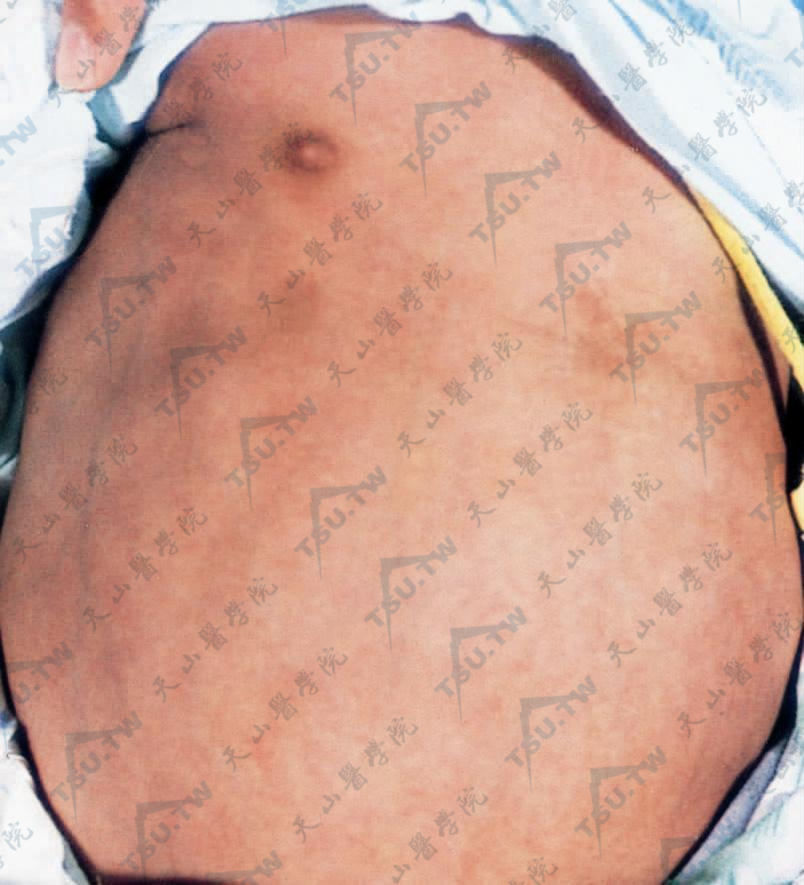
腹部膨隆,皮下脂肪萎缩,皮肤菲薄,腹壁静脉清晰可见
有的病例血浆中促肾上腺皮质激素释放因子、卵泡刺激素释放因子和黑素细胞刺激素释放因子的水平升高,推测为神经功能异常所致。糖耐量降低是该病特征性表现,有高胰岛素血症,糖尿病胰岛素抵抗在早期就可测到,但无危及生命的酮症酸中毒。高甘油三酯血症大部分因血糖升高造成。腹部CT扫描和超声波检查可发现内脏周围脂肪减少,肝脏脂肪增加。脑部CT扫描可见脑室增大,X线检查有灶性骨损害(骨硬化及囊肿),MRI检查发现这种骨损害是由于骨髓脂肪缺损所造成。
组织病理:皮下脂肪和内脏脂肪萎缩、消失。但有报道真皮增厚,甚至有硬皮病样改变。
诊断治疗
诊断:本病应与矮妖精貌(leprechaunism)综合征鉴别,后者也有泛发性皮下脂肪减少或缺失,皮肤有皱纹、松弛,黑棘皮病、多毛、腔口部位皮肤褶皱、角化过度、甲营养不良、厚唇及牙龈增生。也有胰岛素抵抗,但无性器官肥大或肝病。有肌肉废用性萎缩、骨龄推迟、生长迟缓,常早年死亡。
治疗困难,目前尚无法恢复失去的脂肪细胞或预防长期糖尿病引起的并发症。芬氟拉明(fenfluramine)2mg/(kg·d)可降低高代谢状态,抑制食欲,对皮肤症状有缓解作用,但远期疗效不明。少食多餐可不断补充热量(患者无脂肪组织储存能量)。用胰岛素样生长因子-1可降低高血糖症和高胰岛素血症。个例报道用依曲替酯治疗本病并发的黑棘皮病有效。
