
巨细胞动脉炎(Giant Cell Arteritis)本病又称为颞动脉炎(temporal arteritis)、颅动脉炎(cranial arteritis)、老年性巨细胞动脉炎(giant cell arteritis of the elderly)、Horton病(Horton's disease)。为有肉芽肿和巨细胞的全层动脉炎,可累及任何中等大动脉、大动脉,局限性或弥漫性炎症可呈节段性累及。常见于50岁以上女性,男女之比是1:(3~4),随年龄增大发病率升高,可发生于任何人种。
病因及发病机制
病因仍不明。可能的病因包括细小病毒19、肺炎衣原体、水痘病毒及其他的人疱疹病毒感染。直接免疫荧光检查示动脉壁中有免疫复合物沉积,说明体液免疫也可能起某些作用。免疫反应的靶位是平滑肌、DNA、中性粒细胞、心磷脂等。
在有炎症的动脉外膜中,能产生IFN-γ的T淋巴细胞可能在该病的发生中起作用。巨噬细胞可能是组织损伤的最终的效应细胞,形成明显的肉芽肿反应,在外膜引起前炎症反应,中膜引起破坏性反应。位于中膜和内膜交界部位的多核巨细胞和巨噬细胞可能受IFN-γ的刺激,产生血小板衍生的生长因子(PDGF)和血管内皮细胞生长因子(VEGF),然后,这些因子刺激引起肌成纤维细胞增生,在动脉内膜下有细胞外基质的沉积,为了支持新增生的内膜,有新血管的形成。巨噬细胞也可产生活性氧(ROS)和基质金属蛋白酶-2(MMP-2,matrix metalloproteinase-2),这些物质能溶解动脉的内弹力膜,随之形成一条肌成纤维细胞移行到内皮下(subendothelium)的通道,造成动脉的内膜增生,最终动脉腔闭塞。
有单卵双胞胎同患此病和家族发病的报道,大部分研究示与HLA-DR4和HLA-DRB*04等位基因相关。
临床症状
开始全身症状有发热、乏力、头痛、肌痛及关节痛,食欲不振、体重减轻、贫血,90%以上病例血沉快。血沉增快的程度与疾病的活动性相关。最常见的皮肤表现是受累的浅表动脉上方发生疼痛性结节。
本病常引起主动脉的颅外分支的血管炎,很少累及颅内血管。颞动脉受累,头痛限于颞动脉区或放射至颅、面及颌部。在颞动脉、枕动脉及面动脉区可触及皮肤表面发红或发绀的,扭曲肿胀而硬的,有搏动性的疼痛性结节或无脉。局部头皮可因缺血发生坏死,但发生率很低。开始损害可表现为沿着受累动脉区域的带状分布的淤斑,以后出现水疱或大疱,随后发生坏死(下图)。也可出现风团、紫癜、脱发、疼痛性结节和网状青斑。但并非所有以上特征性症状在每个患者都发生。舌动脉受侵犯则舌体触痛、肿胀、发绀、温度低、起水疱,甚至发生坏疽,有颞下颌的疼痛。睫状后动脉及眼动脉分支的动脉炎引起缺血性视神经炎、视网膜或脉络膜缺血,可突然发生单侧或双侧视力丧失(约17%),其中双侧视力丧失约占1/3。约10%的中央视网膜动脉闭塞是由于巨细胞动脉炎引起。其他皮肤外的症状,最常见的是咀嚼或说话时出现张口困难和咀嚼疼痛,在咬肌休息时疼痛消失。还有咽痛、前庭症状、耳痛等。
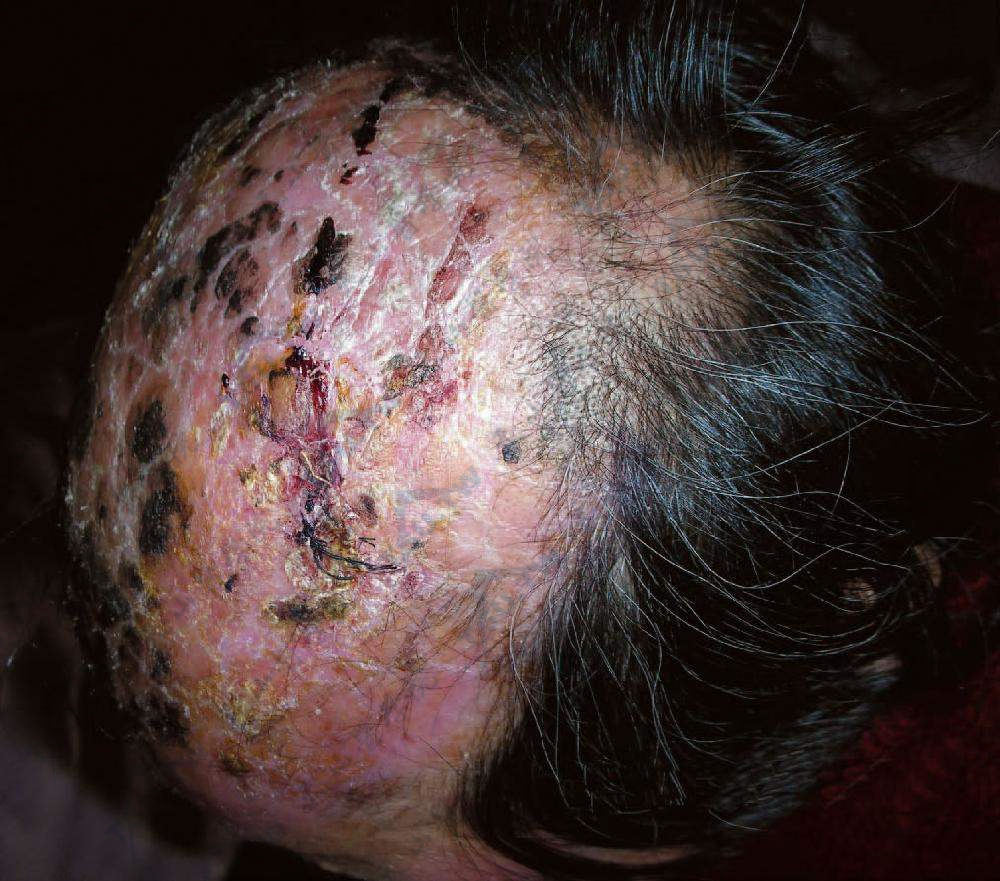
患者头顶及额部头皮因颞动脉炎,局部坏死、结痂,头发大片脱落
颅内动脉受累,可引起包括短暂性局部缺血发作(transient ischaemic attacks)、脑梗死或出现精神症状,但不常见。
仅10%~15%的病例可发生头颈以外的中等或大动脉的肉芽肿性血管炎。可侵犯胸腹部动脉大分支的一部分(包括冠状动脉及肠系膜动脉、子宫动脉),可形成主动脉、颈动脉或其他大动脉的动脉瘤,动脉瘤可破裂。在主动脉弓及其分支部分发生者,临床上很难与多发性大动脉炎相区别。
50%的病例也可有风湿多肌痛(polymyalgia rheumatica)的症状,包括颈、肩和四肢近端肌肉疼痛和僵硬至少有4周,血沉增快,对糖皮质激素反应好。近来认为风湿多肌痛与巨细胞动脉炎是具有相同血管炎疾病的两个不同的疾病。
预后:有眼动脉分支受累者,不管是否用大剂量糖皮质激素治疗,仍可发生失明,一般发生于治疗开始的5天内。本病的死亡与心血管、神经系统及胃肠道血管累及有关。也有报道少数病例可自然缓解。
血常规检查有正色素性贫血,血小板可升高,经糖皮质激素治疗后可很快降至正常。血沉(ESR)加快,大部分超过50mm/h;C反应蛋白(CRP)升高,CRP和ESR同时监测,其敏感性为97%,CRP不受年龄、性别、血浆成分或红细胞形态等因素的影响。
其他的检查包括血黏度、纤维蛋白原、补体、IL-6、α2球蛋白、β球蛋白等可升高。IL-6可能在提示疾病的活动性方面较ESR更敏感。半数患者可有肝功能的异常,尤其是碱性磷酸酶的异常。有一研究显示,抗心磷脂抗体在活检证实的巨细胞动脉炎中阳性率为42%。荧光素血管造影(fluorescein angiography)可见典型的异常改变,包括脉络膜和视网膜中央动脉充盈时间延长,脉络膜动脉无充盈或充盈少。超声波检查为无创伤性检查,用于活检前定位动脉走向,观察眶血流量的改变与病程的关系,受累血管周围有低回声晕(血管壁水肿所造成)。最近有荟萃分析示,低回声晕与颞动脉病理检查比较,敏感性和特异性分别为69%和82%。核磁共振(MRI)可观察到大血管壁增强程度和血管壁的水肿,所以也可用于为活检定位受累血管。正电子发射断层摄影术(positron emission tomography,PET)可观察到大血管的血管炎改变。
组织病理
所有病例均应做受累颞动脉的活检,由于颞动脉炎为“跳跃式”病变(skip lesions),大概跳跃的距离为数mm。所以应取足够长的受累动脉。一般推荐取2cm长,Cohen & Smith提出至少应取2.5cm长。标本除做HE染色外,最好还应做弹力蛋白染色,以了解血管弹力层的破坏情况。虽然该病可侵犯中等大的动脉及大动脉的各层,但主要侵犯的是中膜,表现为慢性肉芽肿性炎症。动脉壁肥厚,内腔因肉芽肿组织增生而狭窄或闭塞。内弹力膜破坏,中膜有明显纤维蛋白样变性,可见多数多核巨细胞浸润,但多核巨细胞并不是诊断所必需的。中膜、外膜还可见淋巴细胞、浆细胞、组织细胞,有时可有嗜酸性粒细胞,可有不同程度的纤维化,故呈闭塞性巨细胞性肉芽肿性全动脉炎的病理表现。可有血栓形成。
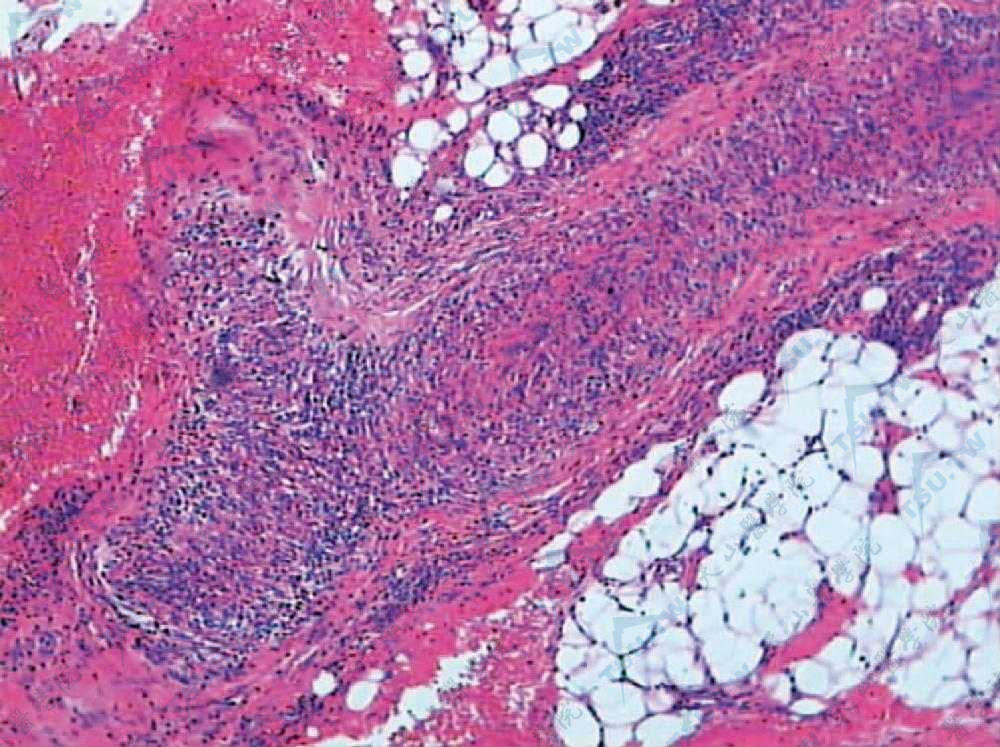
皮下脂肪见一动脉管腔闭塞,血管壁及周围有炎细胞浸润
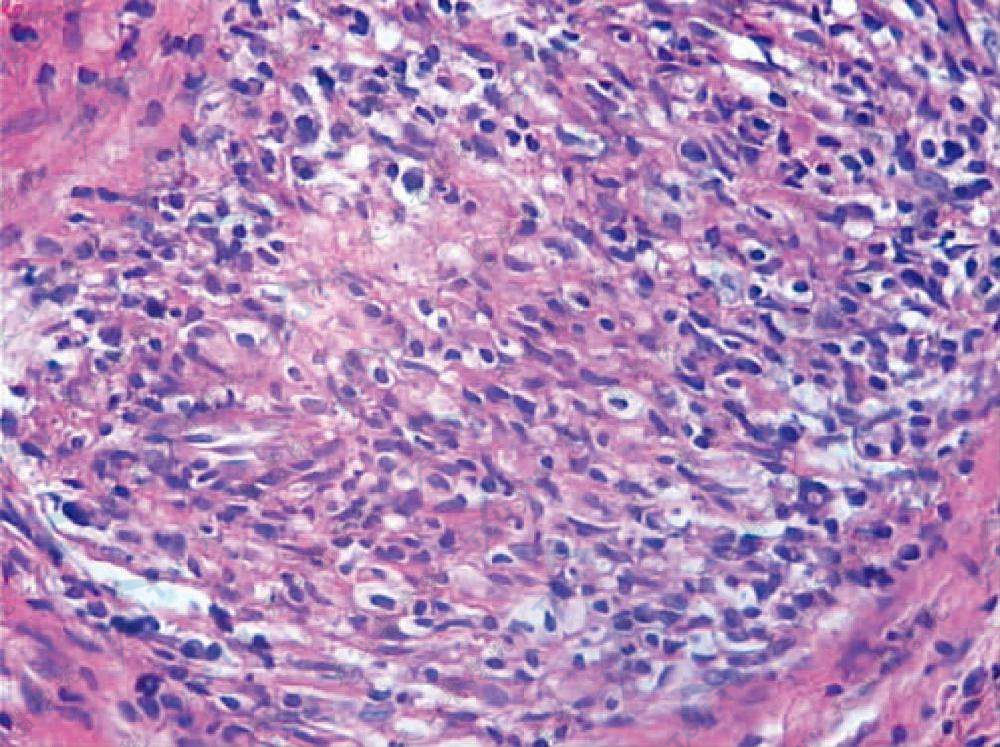
动脉血管壁有组织细胞、淋巴细胞和少量中性粒细胞浸润
诊断及鉴别
巨细胞动脉炎诊断的金标准是颞动脉的组织病理学诊断。磁共振血流成像术(Magnetic resonance angiography)是非创伤性的诊断手段,可帮助证实临床诊断,为活检定最佳部位。1990年美国风湿病学会制订了巨细胞动脉炎诊断标准,出现3个或3个以上以下标准,应诊断为巨细胞动脉炎(敏感性或和特异性分别是93.5%和91.2%):
- 发作时年龄超过50岁;
- 新近发生的头痛;
- 颞动脉异常(疼痛或搏动减弱);
- 血沉增快,用韦斯特格伦法检测,超过50mm/h;
- 受累动脉活检,病理示坏死性血管炎,有单-核细胞浸润为主的或肉芽肿性炎症。
治疗
糖皮质激素的反应很好,是本病的一线治疗药物。泼尼松60~80mg/d(据报道剂量为20~100mg/d),用4~6周。一般系统症状常在治疗后24~72小时内改善,而血沉常需数周才恢复正常。症状缓解后,根据系统症状、ESR和CRP等症状和检查指标缓慢减糖皮质激素量,开始大概每月减10mg/d(或每1~2周减10%),然后每月减5mg/d,当达到10mg/d或15mg/d剂量时,甚至每月仅减1mg/d。或用2.5~20mg/d的维持量,该病为自限性疾病,常在1~2年后缓解,糖皮质激素应用至少1~2年。大部分病例在撤药后可完缓解。如果治疗过程中ESR/CRP升高或症状复发,应警惕疾病的再活动,或维持原糖皮质激素量或增加糖皮质激素用量;ESR/CRP升高还应考虑是否发生了潜在的机会感染。一般在18个月内最易复发,中位易复发的时间为7个月。所以在治疗6~12个月后,可用维持剂量。对于有视觉改变主诉的病例,大部分报道的方法是冲击治疗3天,然后改用2mg/(kg·d)。
如患者有较高水平的循环IL-1β、IL-6和TNF-α则更易复发,应用的糖皮质激素量需较高和较长时间。
为减少糖皮质激素的用量及减少不良反应,可联合用以下药物:甲氨蝶呤、硫唑嘌呤、环磷酰胺(CTX),还有报道可联合用环孢素、氨苯砜和抗TNF-α剂等。有报道抗凝药如肝素治疗可改善视觉,增加眼部血流。
