
本病又称Osler病、Rendu-Osler-Weber病,是以鼻出血和出血、黏膜皮肤毛细血管扩张和内脏动静脉畸形为特征的遗传性疾病。1864年Sutton及1865年Babington均报道了本病,直到Rendu、Osier、Weber才对本病有较完整的描述。
病因及发病机制
属常染色体显性遗传,至少有两个基因突变,即内皮糖蛋白(endoglin简称ENG)位于染色体9q33~34和激活素受体样激酶-1(activin-receptor-like kinase-1,简称ALK1),位于染色体12q13,ENG突变见于HHT1型,ALK1突变见于HHT2型,两者结合到TGF-β超家族,与调节内皮细胞活性和细胞外基质有关,对血管完整性是必需的,在血管生成中起关键作用。ENG表达降低,ALK1和ALK5依赖的TGF-β表达受损,瓦解细胞骨架,不能形成索状结构,血管发育不良,导致小血管变脆伴出血。
Bean认为出血系小动脉生理性收缩功能减退,血管肌肉和弹性组织不能自我完成封闭,止血过程受阻。Hashimoto和Pritzker在电子显微镜下发现血管周围组织缺损、血管内皮细胞间的连接破裂、毛细血管扩张、损害中纤维蛋白溶酶原致活剂成分增多,致毛细血管周围组织的纤维蛋白溶解活性增加而易于出血。
临床症状
皮肤损害始于青春期后,大多30~40岁发病,但广泛皮损亦见于幼儿。皮损可累及任何部位,但以甲床、面、耳、结膜、手、手背、前臂等身体上半部最常见,躯干、股、跖趾很少累及,典型损害为集簇细小扩张的毛细血管丛,呈紫红色或鲜红色小点,单个损害可呈点状、线状、分枝状或蜘蛛样(无搏动性),一般聚合呈斑状或略高起的丘疹,结节性损害罕见。用毛细血管镜检查,见皮内有扭曲扩张的血管团(小血管瘤)或扩张的血管襻。部分病人皮肤易发生淤斑。皮损虽可消退,但倾向持久存在,随年龄增长而增多。
多数病人有黏膜损害,在儿童期较少见,至30岁左右则多见,可累及唇、舌、口腔、腭、鼻中隔黏膜、鼻咽、咽喉、支气管及整个胃肠道,见点状和条状毛细血管扩张,舌部损害有特征性,毛细血管镜下见蕈状乳头内单一明显扩张的血管,并致该乳头增大。
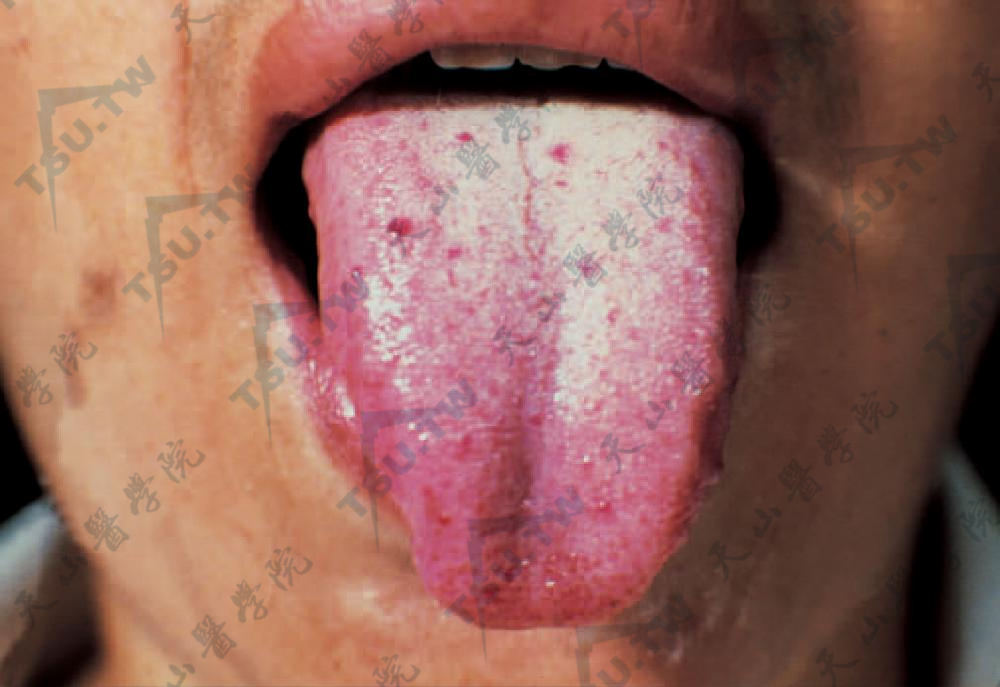
舌黏膜上呈圆点状、边缘整齐的毛细血管扩张。重压之则消退
其他尚可累及球结膜、视网膜、肾、膀胱、肝、脾、脊髓、脑膜和大脑等。肝脏间质组织中,毛细血管扩张伴纤维组织增生,导致肝大和肝硬化,后者与反复输血也有关。
一些病人及其家属中可有大血管畸形,如肺及脑内动静脉瘘、肝动静脉吻合、主动脉和脾动脉动脉瘤、肺动静脉瘘,至青春期可能出现呼吸困难、发绀、杵状指和红细胞增多,脑、脊髓和脑膜血管畸形可产生各种严重的神经症状。
出血可发生于任何部位,但皮肤出血罕见,90%的患者黏膜易发生溃疡和出血,最常见的是反复鼻出血,始于婴儿和儿童,更常见于青壮年,并日趋加重,出血频率和程度影响临床表现和过程,患者可有头昏眼花、心慌、乏力等失血性贫血症状。其他有血便、黑粪、咯血、血尿、月经过多、眼底出血,但均较少见。因血管畸形导致蛛网膜下腔、肝、脾、脑和脑膜出血者极罕见。
实验室检查:毛细血管镜、内镜、动脉造影可见皮肤、胃肠道黏膜小血管扭曲和扩张,出血严重者有严重贫血,肺动静脉瘘者可有红细胞增多。
本病死亡率小于10%,可死于严重并发症,如出血、肺动静脉瘘、脑栓塞、心内膜炎、脑膜脑炎、肺和脑脓肿。
组织病理
真皮乳头层及乳头下层毛细血管和小静脉呈不规则扩张,管壁菲薄,仅有扁平内皮细胞而无周细胞,血管结缔组织变性,电子显微镜检查见扩张的血管为无周细胞的微细静脉。
诊断及鉴别
根据皮肤和黏膜损害,易出血史和有家族史易确诊。本病与Von Willebrand综合征鉴别的依据是后者无皮损,而有血友病证据。皮损中有蜘蛛痣样损害,但无搏动性,这可与一般蜘蛛痣鉴别。实验室和消化道检查可排除其他出血性疾病和一般消化道出血性疾病。本病亦应与其他原因的毛细血管扩张症鉴别。
治疗
治疗原则是控制出血和纠正贫血,可用局部压迫、电灼、透热疗法、激光等控制和治疗个别出血性损害。雌三醇鼻软膏外用可治疗鼻出血,雌激素的止血作用是促进黏膜角化,并非直接作用于血管本身,糖皮质激素亦可减轻出血倾向。有主张切除出血部位鼻中隔黏膜,用分层皮肤移植片植皮,以求止血。用氨基己酸作抗纤溶治疗亦有帮助。肺动静脉和其他部位动静脉畸形可作结扎、切除或介入栓塞治疗。严重贫血者应补充铁剂或输血。
