
本病少见,1931年由Hopf首先描述。
病因
本病是一种常染色体显性遗传性疾病,因基因ATPZA2突变造成,常同时伴发毛囊角化病,有时可见在同一家族的不同成员中分别患有这两种病。Waisman认为本病与掌跖点状角化及毛囊角化病是一种共存的三联征,与同一显性基因传递有关。付风英等(1989年)报道1例疣状肢端角化症合并有毛囊角化病。但本病也可与毛囊角化病无关。
Love等坚持认为本病是一种独立的疾病。徐彩霞(1990年)报道了家族性疣状肢端角化症3例,在家系四代60人中,有21人患病,各代中均无近亲婚配,均有不同程度的细胞免疫功能低下。
临床症状
多在20岁以前发病,男女均可发生,皮损为多发性角化过度性扁平疣状丘疹,质地坚实,直径1mm至数毫米,暗红褐色或正常肤色,常密集成群,颇似扁平疣,但较之更扁平,且对称发生于肢端的手足背部,也可蔓延至手指屈侧、腕、前臂、肘、膝、掌、跖等部位,但颜面及躯干部一般不被累及,皮疹经摩擦可以发生水疱,一般无自觉症状,也有少数病人的皮疹酷似寻常疣。患者可有掌跖部皮肤弥漫性增厚及甲板增厚、浑浊。发病后皮损逐渐增多,持续终生不消退,已有转变成鳞癌的报道。
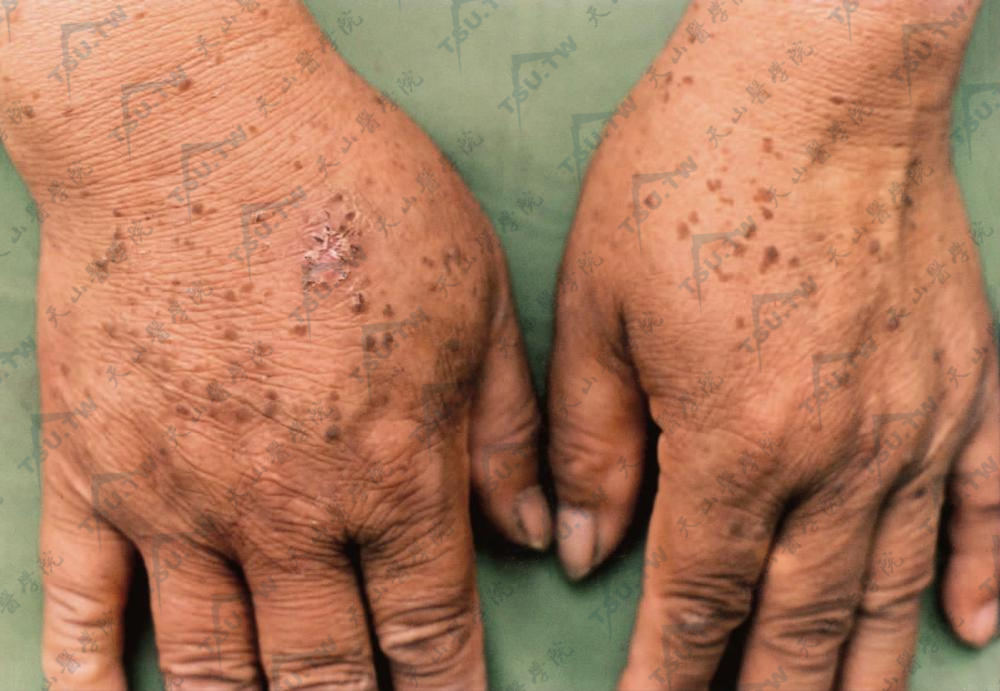
肢端见角化丘疹,棕红或棕色,类似扁平疣,不融合
组织病理
有明显的角化过度,乳头瘤样增生,颗粒层及棘层增厚,但角化不全不明显,表皮呈轻度乳头瘤样增生,乳头顶部隆起如塔尖,表皮嵴轻度下凹,多在同一水平。
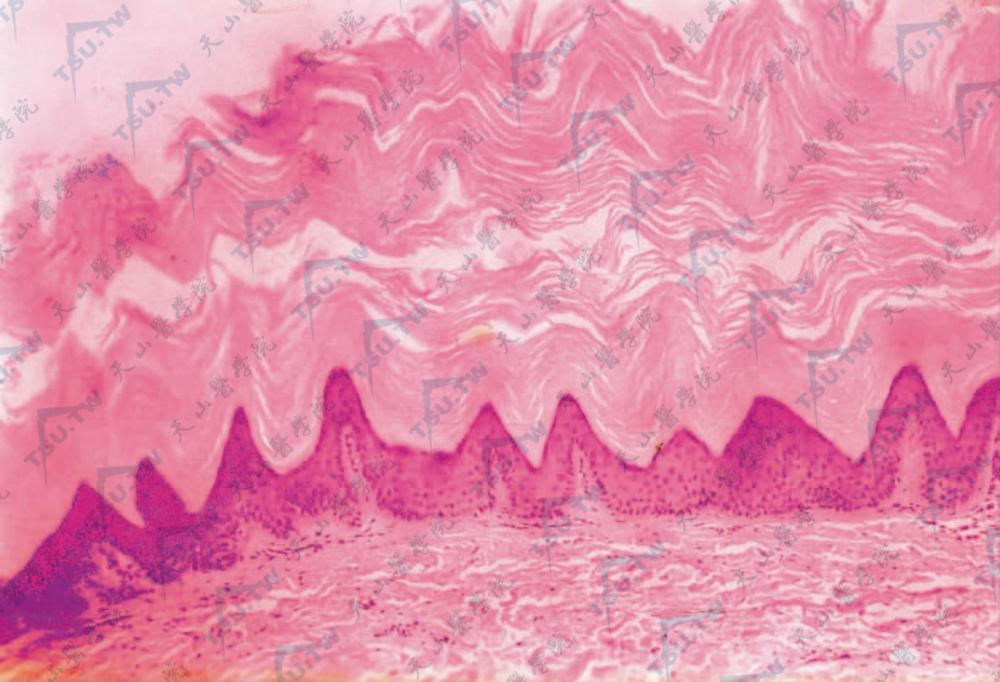
角层高度增厚,表皮山峰状向上增生(HE染色×100)
诊断及鉴别
在肢端尤其在手足背部有多数坚实的角化性疣状扁平丘疹,幼时发病,长期存在以及有明显的家族史等,应考虑为本病,必要时可作病理检查以协助诊断。
需要与下述疾病鉴别:
- 扁平疣:无遗传性,不累及掌跖,发病年龄较晚,病程短,表皮浅层细胞有空泡形成。
- 疣状表皮发育不良:皮损分布广泛,表皮细胞有广泛的空泡形成,核内可发现病毒包涵体。
- 脂溢性角化病和灰泥角化病:发病晚,皮损大,分布部位及病理改变也不同。
- 持久性豆状角化症:组织病理上有马尔匹基层变平和真皮浅层致密的带状细胞浸润。
治疗
目前尚无满意疗法,患者应避免日光曝晒,以免皮损加重或防止诱发恶变。
皮损可试用电灼、CO2激光、液氮冷冻治疗,也可外用0.1%维A酸软膏、5%氟尿嘧啶软膏或作薄层切除,但常易复发。
