
Lichtenstein(1954年)将Letterer-Siwe病、汉德-许拉-克里斯琴综合征(Hand-Schüller-Christian syndrome)和嗜酸性细胞肉芽肿归称为组织细胞增生症X。本病曾称网状细胞肉芽肿、组织细胞肉芽肿、系统性单核巨噬细胞肉芽肿及特发性组织细胞增生症。目前统一称朗格汉斯细胞组织细胞增生症(Langerhans Cell Histiocytosis,简称LCH)。目前将朗格汉斯细胞组织细胞增生症分为:
- 哈希莫托-普里茨克病(Hashimoto-Pritzker disease,HPD)
- 嗜酸性肉芽肿(eosinophilic granuloma,EG)
- 汉德-许拉-克里斯琴病(Hand-Schüuller-Christian disease,HSC)
- 莱特尔-西韦病(Letterer-Siwe disease,LSD)四型。
四型是同一疾病的不同阶段和不同程度的表现,临床上有时可见中间型、混合型或转化过程。因此也可以看做是一个病谱系列的疾病。
病因及发病机制
朗格汉斯细胞组织细胞增生症的病因迄今未明。组织病理提示本病为组织细胞呈新生物状增殖过程,继之发生肉芽肿,最后呈黄瘤样反应。其发病机制有病毒感染、免疫学、细胞遗传学和肿瘤形成假说。本病虽遗传特征尚不清楚,近年来有研究采用X染色体连锁DNA探针和X染色体失活试验表明此病存在克隆性增殖,可能与遗传有关。在同胞兄弟姐妹中的发病率比普通儿童高得多。
近年来大量电子显微镜观察发现本病浸润的细胞中有1/3~1/2的细胞胞质中含有棒状伯贝克颗粒,这种颗粒在正常组织细胞和其他噬脂组织细胞中是没有的,而与表皮中朗格汉斯细胞中的伯贝克颗粒在形态上完全一样。在核的结构和对酶的反应上也均相同,二者都有C3表面受体,在细胞培养中均能吞噬红细胞。免疫组织化学中,S-100蛋白CD1a及花生凝集素均阳性。提示本病是朗格汉斯细胞组织细胞增生性疾病。
另外,该细胞在电子显微镜中可见大小不等的脂质空泡,没有界膜,空泡周围有数个原发性溶酶体,可以认为是该类细胞中聚积的脂质,可能为溶酶体降解产物受损所致。
临床症状
一、Hashimoto-Pritzker病
为四型中最轻型,呈良性转归,通常发生于新生儿或出生后的几天。初起为单个或多个红褐色结节,或呈鲜红色类似血管瘤的皮损。之后丘疹结节可溃烂,皮损的数目和范围可在几周内逐渐增加,之后结痂。病情有自愈性,常在2~3个月内好转。
二、嗜酸性肉芽肿
为四型中较轻型。发病年龄为2~5岁,较大儿童或青年偶见。常为单个或多发性骨损害,X线检查具有特征性的穿凿性破坏区,有时面积可以很大,且易发生自发性骨折。除个别患者出现尿崩症外,一般都无明显自觉症状。本型皮疹少见,为黄色或棕色小丘疹,基本上与其他二型所见相似。头皮可出现斑状炎症损害,类似脂溢性皮炎。偶于外生殖器发生溃疡性肉芽肿,深部肉芽肿可直接向外穿破形成窦道或瘘管。病情进展缓慢。
三、Hand-Schüuller-Christian病
为四型中较重型。本型虽也可发生于婴儿和成人,但常于2~6岁间起病。典型的三联征为颅骨缺损、眼球凸出和尿崩症。不到晚期,三联症状不一定完全出现。本型约30%病人有皮疹。皮疹有3种形态,最常见的为浸润性斑块,可形成溃疡,尤其好发于腋下和会阴及口腔。这种皮疹在骨嗜酸性细胞肉芽肿中也可见到。第二种为广泛而融合的丘疹,上附鳞屑或结痂,好发于头面、躯干或臀部,似脂溢性皮炎样。这类皮疹在Letterer-Siwe病也能见到。第三种为散在的似发疹样黄瘤状黄色丘疹,质软。若单一出现该类皮疹时,无论从临床或组织病理学上都很难与幼年性黄色肉芽肿相鉴别。本型患者常无系统自觉症状,但却可发现多系统体征。
最常见者为骨受侵,特别是颅骨,在颅骨缺损处常可扪到结节,X线检查更能明确。其他下颌出牙处的骨质侵蚀,常见牙龈溃疡。1/3患者常有肺门及肺中心区弥漫性浸润,但不累及周边部位。肝脾及全身淋巴结常肿大,甚至出现肝内阻塞性黄疸。有些病人以慢性中耳炎就诊,通过X线检查发现乳突和颞骨岩部的病变而诊断本病。眼球凸出为本型三大症候之一,约10%患者出现本症候。由于脑垂体茎部或下视丘受累,约50%患者发生尿崩症。患有贫血者少见。
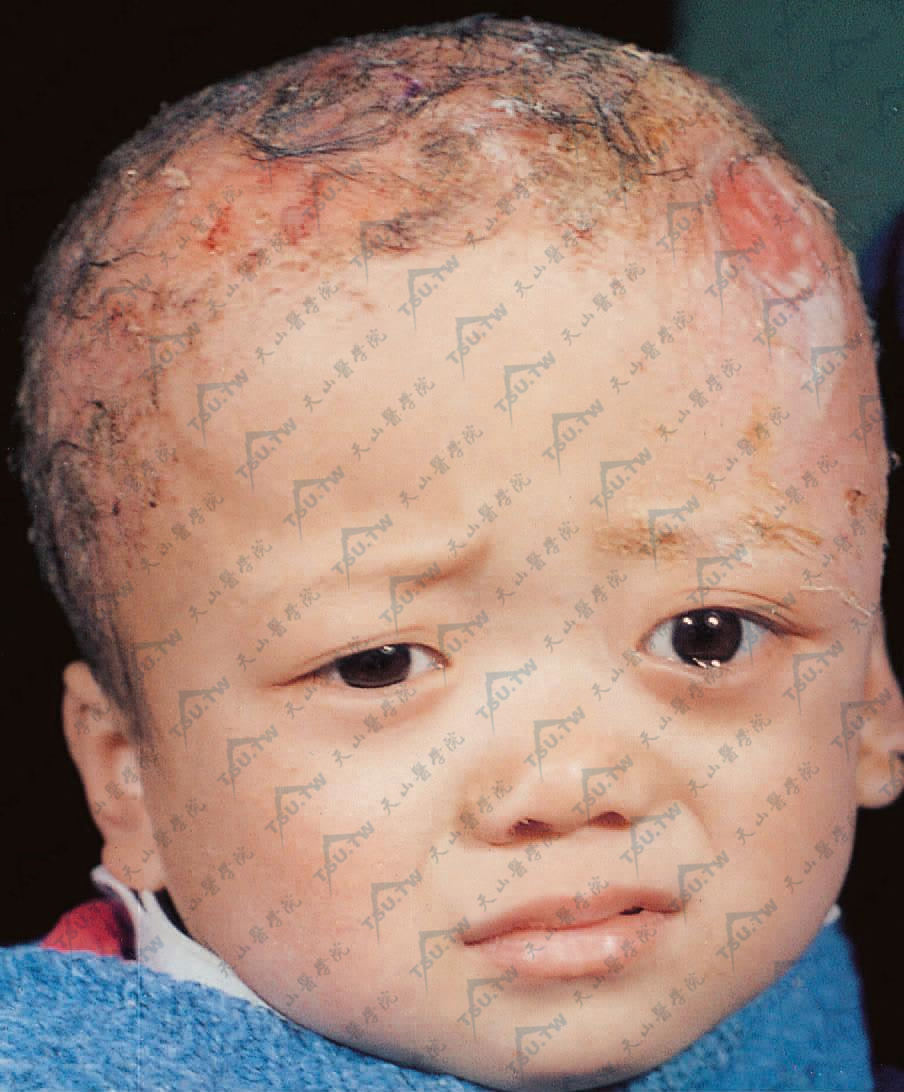
头部广泛融合的丘疹,附鳞屑,似脂溢性皮炎。左眼球凸出,左颞溃疡。X线颅片示下方颅骨缺损
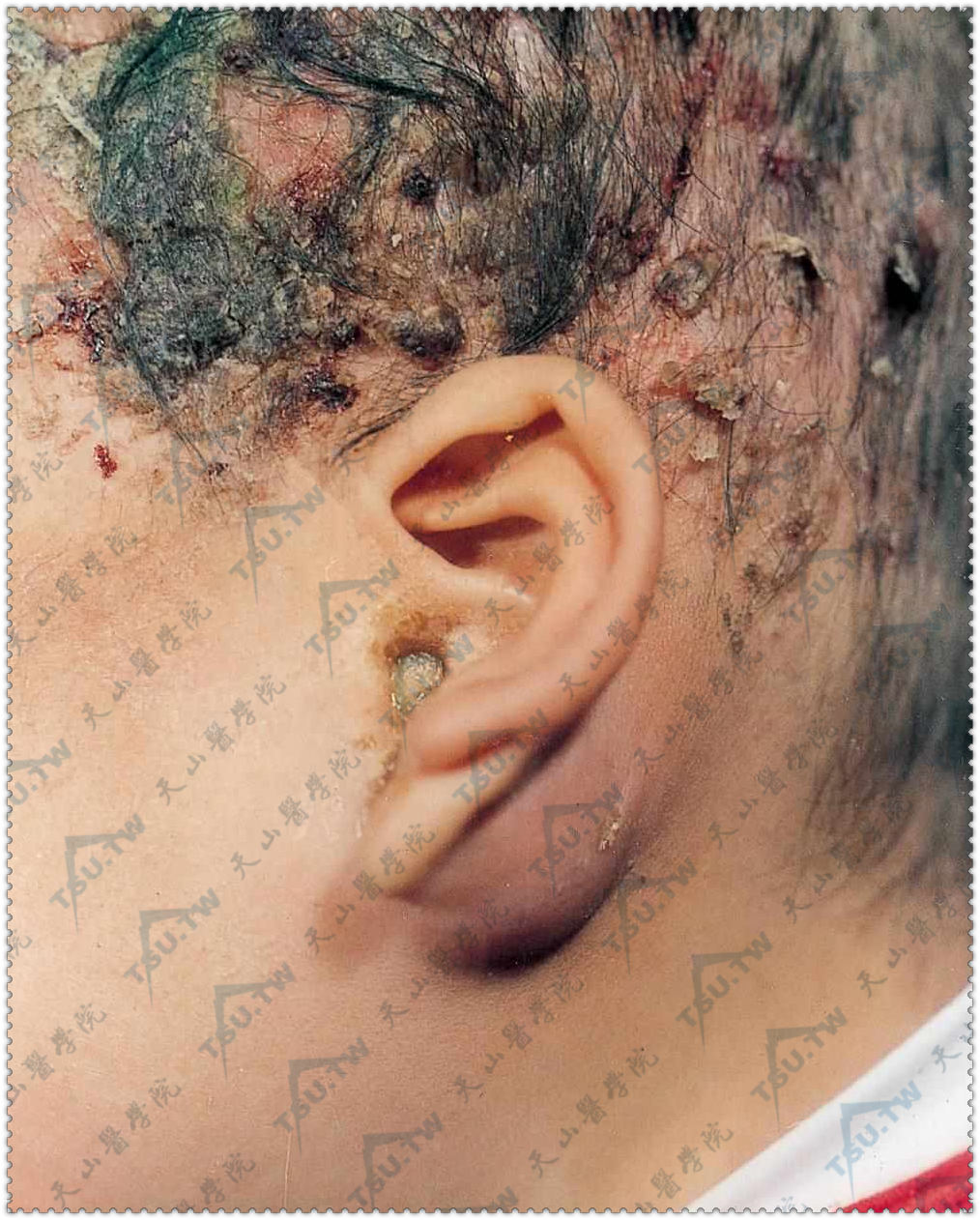
头部脂溢性皮炎样皮损,左耳后乳突部红肿,外耳道溢脓
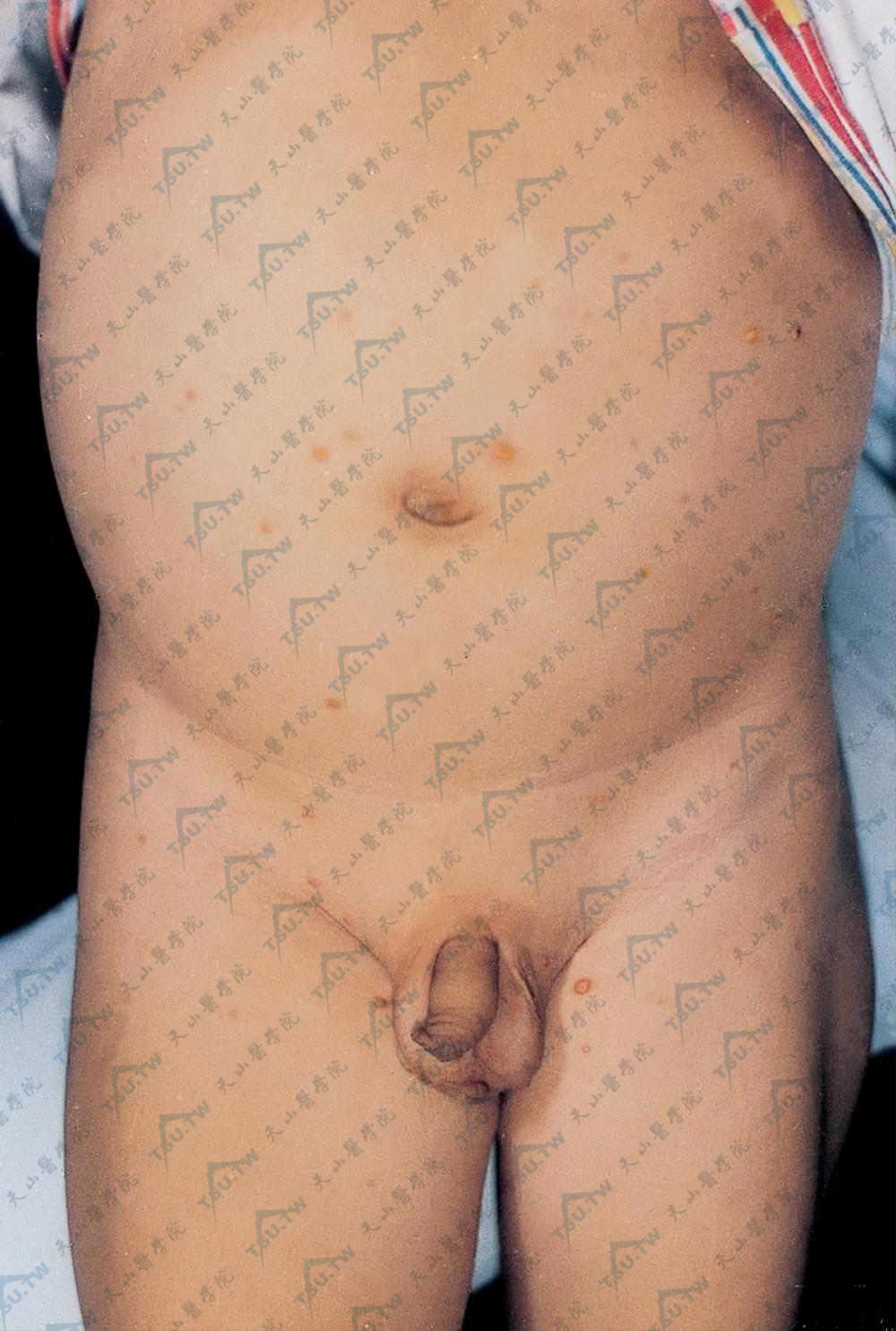
腹部、外生殖器附近散在黄红色丘疹、小结节,质软,似幼年性黄色肉芽肿
四、Letterer-Siwe病
为四型中最重型。常发生于婴儿,多在9个月内发病。约80%病例有皮损。典型皮疹具有诊断性,表现为群集的黄棕色鳞屑性斑丘疹,分布广泛,常见于头、面、颈、躯干和臀部。躯干皮疹可呈紫癜状。口腔黏膜可见坏死或肥厚性损害。此外,皮肤尚可出现结节或溃疡,有时头皮、躯干皮疹与脂溢性皮炎或毛囊角化病很相像。偶尔皮疹仅为紫癜性。本型早期可有系统症状,如体重减轻、乏力和少数不典型皮疹。起病急性者可有发热、肝脾大、淋巴结肿大、贫血和典型皮疹。约10%患者周围血中嗜酸性粒细胞增高。有报道早期即发现有幼稚白细胞者。X线胸片检查可发现多发性肺囊肿,主要表现为粟粒状斑点,偶尔有骨质缺损。病情进展迅速且伴多器官衰竭,几乎都于几个月至1年内因内脏损害或因抵抗力差并发细菌或病毒感染而死亡,也可因麻疹后并发慢性进行性巨细胞肺炎而死亡。
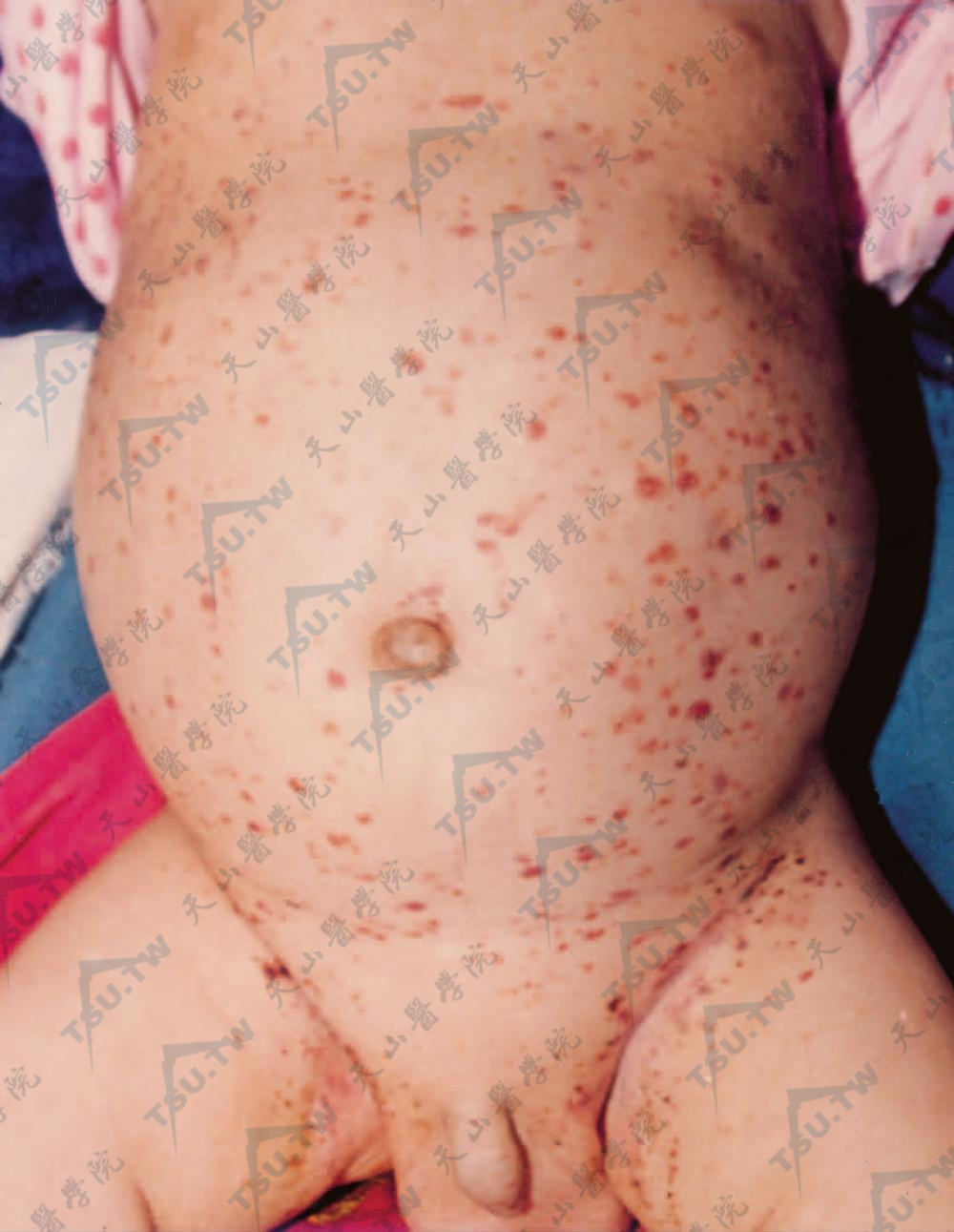
婴儿躯干部发生群集的黄棕色鳞屑性斑丘疹,有的可呈紫癜状,分布广泛
组织病理
由于本病四型在组织病理上均有四个不同时期:增殖、肉芽肿、黄色瘤和纤维化,所以相互间容易混淆,但组织病理仍是诊断本病的主要依据。
Letterer-Siwe病几乎全为增生的组织细胞所浸润。这种细胞较大,胞质清晰,细胞境界清楚,呈水肿样疏松排列,常为单一型,但也可为不典型的小核,或细胞大小不等,许多核可呈肾形,核丝分裂不常见,核仁不清楚。胞质深染,有时含有颗粒或细胞碎片,有些胞质中含脂质呈泡沫化。
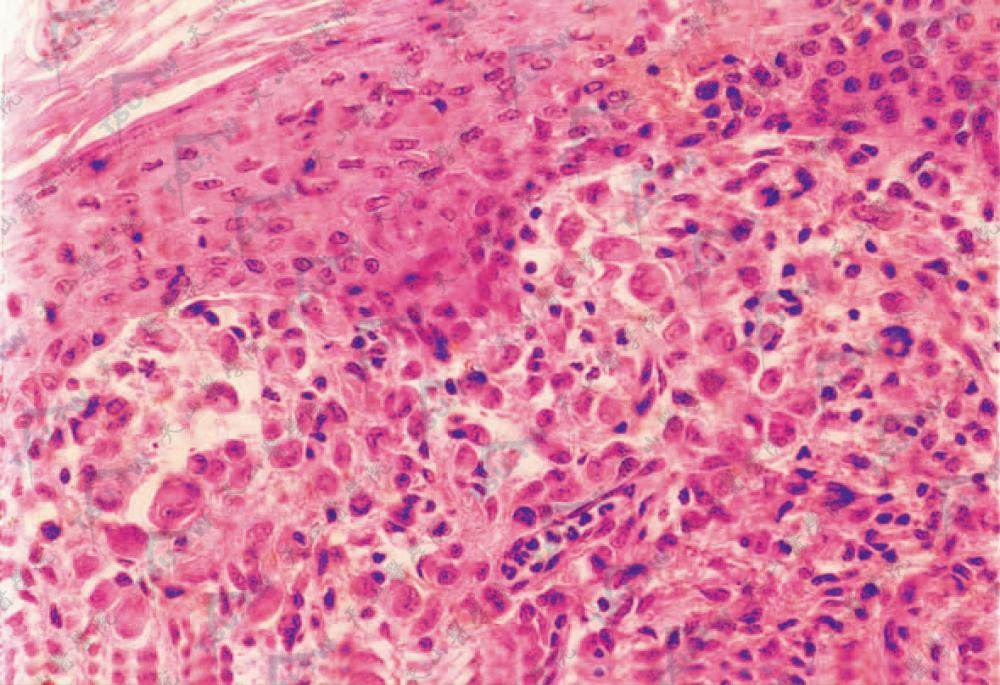
浸润的组织细胞胞体圆大,胞质丰富,淡伊红色,侵及表皮(HE染色×400)
Hand-Schüller-Christian病和嗜酸性细胞肉芽肿中主要表现为非特异性炎细胞浸润。组织细胞增生也能见到,但大部分被炎细胞所掩盖。当损害消退或痊愈时,则主要表现为纤维化。
慢性型成熟损害中,可见大量嗜酸性粒细胞。这些细胞的出现或增多对诊断并无重要意义,免疫病理标记,常阳性表达S-100蛋白、CD1a等。
诊断与治疗
四型都可结合组织学特点而诊断。皮疹应注意与脂溢性皮炎相鉴别。骨损害应与多发性骨髓瘤相区别。
本病目前还无满意疗效。Letterer-Siwe病应用糖皮质激素、抗叶酸药物和烃化剂治疗,能缓解病情或转为慢性型。有报道单用长春花碱获显效。通常以联合化疗疗效较佳,但应密切注意危险的不良反应。对Hand-Schüller-Christian病和嗜酸性细胞肉芽肿的骨损害可采用放射治疗,单个损害可用外科刮除。内脏受侵者也可用抗叶酸药物治疗。
